
Программа для построения 3д моделей молекул органических веществ
Обновлено: 02.07.2024
В данной работе, рассматривались вопросы, связанные с 3D визуализация пространственных молекул органических соединений с помощью ACD / 3D VIEWER. Главной целью нашего исследования было изучить, перевести и создать брошюру-инструкцию для учащихся и студентов по использованию данного приложения. В нашей работе рассмотрены такие вопросы:
Какое программное обеспечение существует на данный момент по визуализации органический соединений?
Что такое ACDLabs ?
Что необходимо знать, чтобы уметь пользоваться 3 D Viewer ?
Как упростить работу по использовании ACDLabs для студентов и учащихся?
Нами была проанализированная специализированная литература и интернет ресурсы по данной тематике. В заключении приведены обоснованные выводы, сделанные на основании проделанной экспериментальной работы
Глава 1. Программный пакет ACDLabs…………….……………………. ………6
1.1 ACD/ChemSketch и ACD / 3D VIEWER…………..………………….…………6
1.2 Алгоритм 3 D -оптимизации…………………………………………….………..8
Глава 2. Работа ACD/ChemSketch и ACD / 3D VIEWER………………………. 10
2.1 Инструкция по работе с ACD/ChemSketch………………………..………..…10
2.2 Установка и файловый ассоциации 3D VIEWER……………………………13
Глава 3. Экспериментальная часть………………………………………………. 16
3.1 3D оптимизация молекул………………………………………………………16
3.2 Настройка изображения молекулы…………………………………………….18
3.3 Вычисление и изменение структурных параметров………………………….20
Создание и совершенствование компьютерной техники, появление новых технологий передачи, обработки, накопления и представления информации облегчило и ускорило вычислительную работу во многих областях науки и, в частности, в химии, Квантовая химия, молекулярная механика, планирование химического синтеза, получение и обработка экспериментальных данных с помощью новых информационных технологий и компьютерной техники — это лишь некоторые типичные примеры. Увеличение вклада информационных технологий в развитие химии связано, в первую очередь, с появлением персональных компьютеров с высокими эксплуатационными характеристиками и новых технологий передачи информации. Благодаря их относительно низкой цене и доступности для широкого круга пользователей такое оборудование стало обычным в большинстве химических лабораторий и учебных заведениях мира.
Одной из сфер использования компьютеров среди химиков является подготовка текстов, содержащих химические структуры. Популярные текстовые редакторы, например Microsoft Word и др, несмотря на свои широкие возможности, не имеют удобных инструментов для создания химических структур высокого качества. Для их создания и редактирования имеется целый ряд программных решений, с некоторыми мы и познакомимся в нашей работе. Они различаются по специализации и своим возможностям, по степени сложности интерфейса и работы в них и т.д.
В связи с большой распространенностью программ, работающих в ОС Windows, в настоящем пособии внимание уделено только программам, работающим под управлением данной операционной системы. При этом следует иметь в виду, что существуют как версии рассмотренных программ для других операционных систем, так и самостоятельные программные продукты. Отдельную область представляют собой онлайновые химические редакторы. В нашей работе дается достаточно полное описание химического редактора ACD / 3 D VIEWER .
Актуальность нашей работы заключается в том, что использование технических средств для изучения движения и взаимодействия веществ на атомно—молекулярном уровне, упрощает понимания о реактивной способности соединений. В настоящее время большинство реально эффективных и действующих программ для химии, создаются за рубежом, их понимание и осваивание в первую очередь затрудняется тем, что данные программы выпускаются без дополнительных «языков», и существуют только на языке их создателей (чаще английском).
Целью нашей работы было : знакомство с некоторыми основными видами программного обеспечения, используемого в области химии для рисования структурных формул и 3D визуализации молекулярной структуры, которая дает возможность оценить взаимное расположение атомов молекулы в пространстве, найти межатомные расстояния и валентные углы позволяет решать многие задачи строения и реакционной способности молекул.
Гипотеза исследования: если существуют удобные англоязычные программы для построения органических соединений, что можно предпринять, чтобы упростить их понимания для российских учащихся, студентов и ученых.
Нами были поставлены следующие задачи:
Изучить методическую литературу и пособия по построение органических соединений;
Изучить и выявить более удобную и легкую в использование программа для построения органических соединений;
Научиться пользоваться, и понять технологию использования зарубежных программ по построение молекул органических соединений;
Составить краткое пособие по использованию ACD / 3D VIEWER.
Глава 1. Программный пакет ACD Labs
ACD/ChemSketch и ACD / 3D VIEWER
Advanced ChemSketch Development Inc . - это канадская компании разрабатывающая программное обеспечение ACDLabs для ученых и специалистов в области химических, биохимических и фармацевтических исследований основана выпускниками Московского государственного университета. Область деятельности компании лежит в создании программ для рутинной работы. Анализа предсказывания свойств и управления уже полученными результатами спектроскопических, хромато графических и других методов инструментального анализа химических веществ. Также программный комплекс ACD/Labs известен благодаря возможности предсказывать такие физико-химические свойства. как pKa и LogD и умению генерировать название вещества в различных видах номенклатур. Для построения и визуализации структурных формул предназначены компоненты пакета:
ChemSketch - молекулярный редактор двумерных химических структур и графический редактор;
3D Viewer — программа моделирования и визуализации трехмерных структур.
Химический редактор ChemSketch ориентирован на работу с органическими формулами среднего уровня сложности (имеется большая библиотека готовых формул), но в нем удобно составлять также химические формулы неорганических веществ. С его помощью можно оптимизировать молекулы в трехмерном пространстве, вычислять расстояния и валентные углы между атомами в молекулярной структуре и многое другое.
Программа ChemSketch содержит и инструменты для создания векторных изображений, во многом аналогичных векторному редактору Microsoft Office, поэтому позволяет создавать графические иллюстрации. Создание сложных формул и рисунков облегчается наличием альбома шаблонов формул и рисунков, который может пополняться пользователем. Созданные с помощью редактора объекты могут быть сохранены, распечатаны, а также скопированы в WORD и другие приложения.
Полезен будет и встроенный калькулятор ChemSketch, позволяющий рассчитывать многие характеристики веществ, формулы которых создаются в редакторе.
ACD/3D Viewer – программа быстрого и точного моделирования и визуализации структур. Она полностью связана с программой ACD/ChemSketch, что позволяет Вам рисовать структуры 2D и быстро получать из них прекрасные цветные 3D изображения. С программой ACD/3D Viewer Вы сможете:
Управлять 3D моделями: перемещать, вращать 2D и 3D изображения, изменять размер, стили и цвета;
Отобразить 3D структуру в виде стержней, стержней и шаров, сфер или дисков;
В 3D структуры твёрдых веществ добавлять изображение границ окутывающих сил Ван-дерВаальса в виде мелких точечек; • Измерять и изменять длину связей, углы между плоскостями связей и торсионных углов;
Оптимизировать структуры, используя силовые поля 3-D CHARMM-типа;
Щелчком кнопки в окне программы ACD/ChemSketch переключаться от 3D дисплея к 2D;
Пользоваться конфигурацией автономного вращения 3D молекулы, с/или без изменения стиля показа структуры;
Рассматривать 3D структуру в перспективе;
Отображать многократные связи в режиме показа вращения проволочных структур (Wireframe);
Экспортировать 3D модели в другие программы геометрических оптимизаций и использовать их как хорошие стартовые конфигурации.
Алгоритм 3D- оптимизации
Алгоритм 3D оптимизации быстро преобразовывает плоскую (2D) структуру созданную в программе ACD/ChemSketch в реалистическую 3D структуру. Это основано на изменении молекулярной механики, которая принимает во внимание длину связей, величину угла, внутреннее вращение и не создающих связей Ван-дер-Ваальсовых взаимодействий. Модификации включают незначительные упрощения потенциальных функций и осуществление схемы минимизации дополнительными эвристическими алгоритмами для «плохо ведущих себя» стартовых конфигураций.
Алгоритм 3D оптимизации – патентная версия молекулярного механизма с силовыми полями, первоначально основанными на CHARMM параметрах.1 Модификации включают некоторые упрощения и были предназначены для увеличения стабильности и скорости вычисления. Обратите внимание, что 3D оптимизация НЕ является полномасштабным молекулярным двигателем. Она скорее стремиться воссоздать как можно ближе действительности соответствующие 2 D конфигурации (возможно очень неблагоразумные), чем точно оптимизировать 3D структуры.
Обратитесь к B.R. Brooks, R.E. Bruccoleri, B.D. Olafson, D.J. States, S. Swaminathan, и M. Karplus. CHARMM: программа для макромолекулярной энергии, минимизации и динамических вычислений. J. Comput. Chem. 4 187–217 (1983).
Иногда 3D оптимизация производит молекулярную конфигурацию отличную оттого, что Вы ожидали. Это отражает саму сущность структурного анализа – молекулы обычно имеют несколько возможных конфигураций. Оптимизатор находит только одну и она не обязательно та, которую Вы ожидали. Например, Вы, вероятно, ожидаете, что фрагмент циклогексана будет иметь форму кресла, но оптимизатор может произвести искривлённую форму - лодки, которая так же является одной из возможных конфигураций (в действительности этот фрагмент существует в искривлённой форме во многих структурах). Чтобы получить другую конфигурацию в полученной 3D структуре переместите некоторые её атомы, чтобы сделать начальную структуру ближе к окончательной конфигурации и затем оптимизируйте структуру ещё раз. Если Вы пробуете получить в процессе оптимизации определённый энантиомер для структур с хиральными центрами, вводимая конфигурация может измениться к одной из противоположных форм. Чтобы решить эту проблему, обычно достаточно рисуя поменять местами все четыре хиральных углеродных атома и установить нужное направление связей в начальной 2D структуре, для этого используя оба инструмента Up (Вверх) и Down Stereo Bond (Вниз Стерео связи). Если это не поможет, Вы можете вручную переместить атомы в заключительной 3D формуле и оптимизировать структуру ещё раз. В любом случаи мы рекомендуем ответить «Нет», когда Вас спросят в открывающемся диалоговом окне, о том хотите ли Вы удалить водородные атомы перед начальной оптимизацией.
ACD/3D Viewer программа может оптимизировать только структуры содержащие атомы от водорода до ксенона со стандартной валентностью и в связанных состояниях. Также она не принимает во внимание водородные связи.
В структуре молекулы подвергающейся оптимизированию не может быть более 250 атомов, включая и число невидимых водородных атомов.
Глава 2. Работа ACD/ChemSketch и ACD / 3D VIEWER
2.1 Инструкция по работе с ACD/ChemSketch
Установите и запустите программу ChemSketch 12. В бесплатной (ограниченной) версии при запуске показывается рекламное окошко и не работает часть функций программы (генерация названий по формуле для веществ с числом атомов более 50, доступ к он-лайн базам данных). Все функции рисования рабочие. Результат может быть сохранен в собственном формате программы, нескольких вариантах программ для рисования формул или в виде изображений. Программа интегрируется с MS Word и позволяет вставлять формулы в документы Word.
Для рисования простых химических формул щелкните левой клавишей мышки на лист. Появится молекула метана. Щелчок на молекуле метана даст молекулу этана. Щелчок на любом атоме углерода удлиняет в этом месте углеродную цепь на одно углеродное звено. Щелчок на атомах в середине цепи создает разветвления углеродной цепи. Щелчок на связи между атомами увеличивает ее кратность.
Для исправления ошибочно введенных звеньев есть несколько инструментов. Стрелка Undo Draw на панели инструментов отменяет последнее действие. Справа от нее есть инструмент Delete, с помощью которого можно удалить ненужный атом или связь в формуле. Для выделения и перемещения отдельных атомов используется инструмент Select/Move на панели инструментов. С его помощью можно выделить группу атомов и нажав клавишу Delete удалить их. Для вращения выделенных атомов или групп атомов, используется инструмент Select/Rotate/Resize.
Для ввода атомов, отличных от углерода, используется панель слева от листа. Выбрав на ней нужный элемент (если его нет в списке, его можно найти в таблице Менделеева наверху левой панели), можно этим элементом заменить атомы углерода в уже нарисованной углеродной цепи. При этом элемент подставляется в обычной для него валентности. Для изменения обычной валентности на другую, требуется дважды щелкнуть на элементе при нажатом инструменте Select/Move, нажать на значок V в открывшемся окне свойств атома и выбрать валентность в списке справа.
Для упрощения рисования циклов и некоторых радикалов, справа от листа есть панель радикалов. Список радикалов вызывается щелчком на значке вверху панели.
Для рисования сложных структур в программе предусмотрены образцы формул. Они могут быть вызваны через меню (Templates – Templates Window) или значком Open Templates Window на панели инструментов. В левом выпадающем списке перечислены группы веществ, в правом – страницы и число страниц с примерами. Для использования любого примера достаточно щелкнуть на него левой клавишей мышки и затем щелкнуть в любом месте листа.
Для ввода в структуру радикалов, цифр и т.п., не предусмотренных программой используется функция Edit Atom Label на левой панели.
Для рисования ионов, на левой панели есть инструмент со знаком +. Нажав на светлый треугольник внизу иконки получаем список знаков для ионов, радикалов и ион-радикалов. С помощью этого инструмента можно изменить (или убрать) заряд ионов.
Для отображения атомов углерода в середине цепи (по умолчанию они обозначены графами), выделяется необходимая часть молекулы, в меню вызывается Tools – Structure Properties и на вкладке Atom ставиться галочка Show.
Для симметричного отображения структуры нарисованной от руки можно воспользоваться инструментом Clean Structure. В этом случае выделенная часть структуры (если ничего не выделено – то вся структура) будут приведены к более симметричному виду. Следует иметь ввиду, что функция часто не пригодна для выравнивания полициклических или сильно разветвленных структур, но ей можно выравнивать не только всю структуру, а также выделенную часть молекулы.
В программе можно рисовать схемы химических реакций, химические установки и т.п. Для этого есть соответствующие инструменты на панели инструментов и некоторые примеры в образцах формул. Маленький светлый треугольник на некоторых инструментах открывает список вариантов инструмента.
Для оформления схем, рисунков и т.п. можно переключится в режим Draw на панели инструментов.
В правом нижнем углу программы имеется иконка Properties, позволяющая рассчитать некоторые физические свойства для нарисованной структуры.
Программа позволяет автоматически нумеровать положения нарисованной структуры. Для этого служит команда в меню Tools - Auto Renumbering. Команда Tools - Clear Numbering убирает нумерацию, если она есть.
Для сохранения нарисованной структуры можно пользоваться командами File - Save as. и File - Export.
Шикарный молекулярный редактор с кучей настроек. Да, англоязычный, но это не проблема.
Можно не только нарисовать сложную молекулу в 2D, но и увидеть ее пространственную структуру в 3D (можно повертеть и так и сяк).
Можно отобразить дипольные моменты, Ван-дер-Ваальсовы сферы, заряды, распределение электронных облаков (визуально как прозрачных, так и непрозрачных), сменить стили прорисовки в 3D и так далее.


Лига Химиков
1.2K постов 10.6K подписчиков
Правила сообщества
Старайтесь выбирать качественный контент и не ставьте теги моё на копипасты
Посты с просьбой решения домашнего задания переносятся в общую ленту
1. Оскорблять пользователей.
2. Постить материал далеко не по теме и непотребный контент (в остальном грамотно используйте теги)
3. Рекламировать сомнительные сайты и услуги коммерческого характера
А там есть функция, типа рисуешь желаемую молекулу, и оно показывает реакцию как её получить? Желательно в домашних условиях без всяких заумных реагентов.
Есть ещё такая рисовалка как ChemDraw 3D. На английском, рисовашка схожа с указанной программой, но функционал больше-название и информация о нарисованном соединении (молярная масса, длины связей). Также можно на ней поставить сферический фактор, ЯМР посмотреть, и по названию соединение нарисовать.Я хоть и не химик и вообще в химии нихера не понимаю, но подтверждаю, прикольный инструмент. Может химики и знающие люди подскажут, может нечто созданное мной существовать в природе?
Сильно неплоские молекулы в ней невозможно нарисовать, увы.
А электронные оболочки эта штука может рисовать? Они так то вообще не круглые. В случае сложнее водорода.
Комментарий удален. Причина: данный аккаунт был удалёнСкинул бы кто еще ссыль на работающий с Windows 8 ADVASP.
А для документов ChemDraw само оно
Я нисколько не химик, но я очень надеюсь, что эта программка не врет. Очень. Надеюсь.зы. я программист :-)
Формула зеленоградской воды
В городе Зеленоградск (Калининградская область) мне на глаза попался питьевой фонтанчик с вот такой очаровательной табличкой:

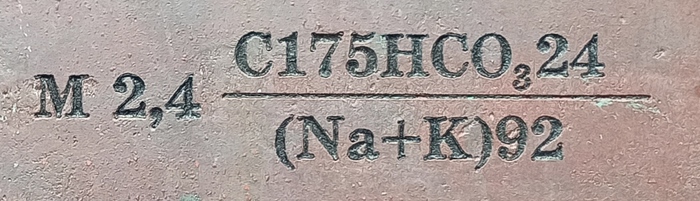
UPD: благодаря пикабушникам сей ребус был разгадан. Подразумевалось следующее:
Общая минерализация 2,4 г/л
Из них хлориды 75%, гидрокарбонаты 24%
Содержание натрия и калия 92% от всех катионов
Самая ненавистная греческая буква




Когда прогуливал химию

Таблица цветовых обозначений элементов в 3D моделях молекул
Пособие для химиков-дизайнеров:

P.S. примеры моделей:
Диоксид углерода, иодоводород, трифторид алюминия и серная кислота.
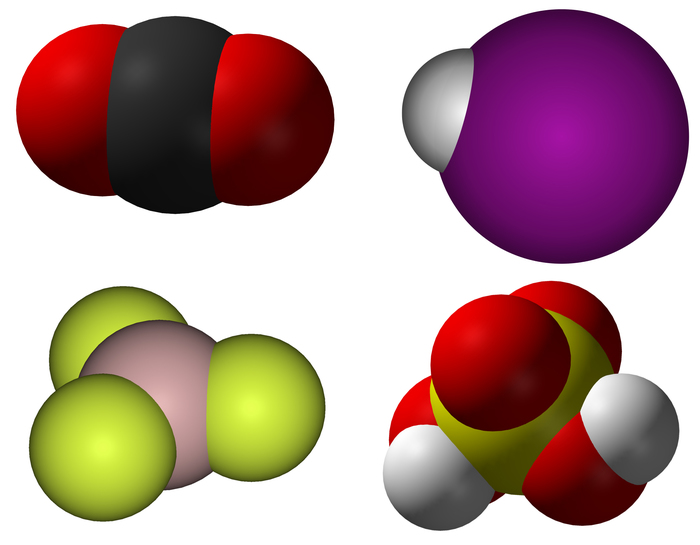
Модели структур различных веществ
Диоксид кремния, SiO2 (кварц)
Фиолетовый - кремний, белый - кислород.

Оксид алюминия, Al2O3 (корунд)
Красный - алюминий, белый - кислород.
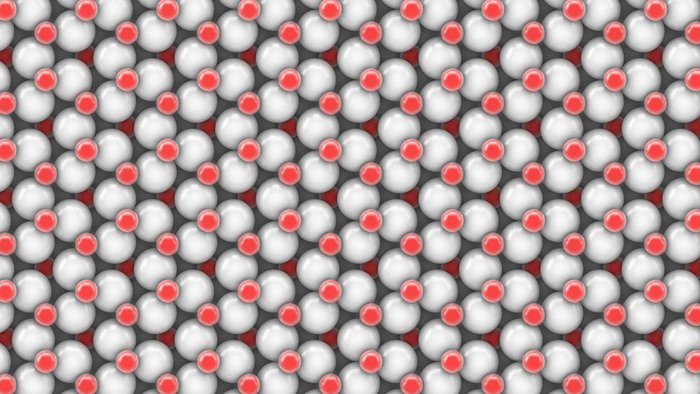
Диоксид углерода, CO2 (сухой лёд)
Черный - углерод, белый - кислород.

Хлорид натрия, NaCl (поваренная соль)
Голубой - натрий, белый - хлор.

Вода (жидкая), H2O
Белый - водород, серый - кислород.

Nd2Fe14B (неодимовый магнит)
Оранжевый - неодим, голубой - железо, белый - бор.

Углерод, C (алмаз).



Источник BeautifulChemistry
Формула борща
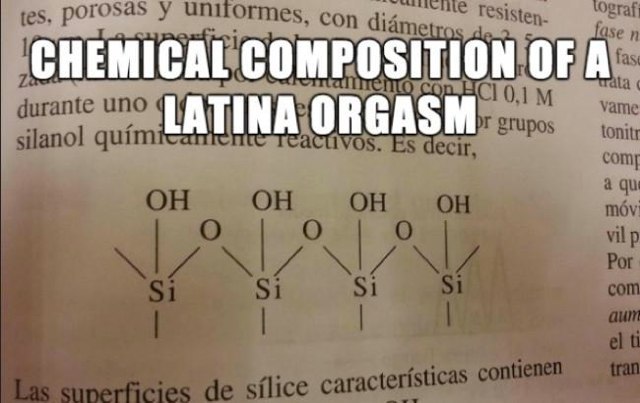
Формула латиноамериканского оргазма
Чем горжусь и первый печальный опыт )) Идея подарка для химиков, медиков и просто любимых)
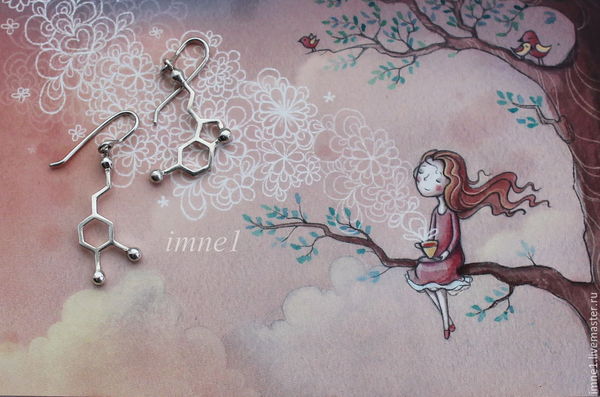
Крутая идея с запада, мое исполнение ))
Почему крутая? Потому что помним:

Как-то попросила меня знакомая сделать кулончик-серотонинчик ее подруге ))
Я сделала нечто, очень похожее на тушканчика и очень этому радовалась. Выглядело это так:

Потом я сделала еще дофаминчик, и выглядел он немногим лучше:

Не, по правде говоря, на деле они были блестящие и симпатичные, но сейчас смотреть на них смешно, потому что теперь я делаю их так:


Если вкратце о молекулах (химики эту инфу, конечно, покритикуют, но надо же как-то объяснить):
Гормон дофамин относится к группе так называемых гормонов радости, удовольствия, веселья и эйфории. Кроме того, именно гормон дофамин толкает нас на подвиги, безумства, открытия и свершения. Когда мы испытываем недостаток дофамина в организме — мы становимся унылыми ипохондриками, тогда как высокий уровень этого гормона превращает нас в донкихотов и оптимистов.

Все программы для молекулярного моделирования делятся, грубо говоря, на две категории: вычислительные пакеты и ПО для визуализации и анализа. Первые - это команднострочные программы, которые решают классические уравнения движения атомов в системе (молекулярная динамика) либо рассчитывают электронную структуру молекул на квантовом уровне (квантовая химия). Эти программы обычно устанавливаются на специализированных вычислительных кластерах и выполняются параллельно на нескольких процессорах (от нескольких штук до тысяч в зависимости от задачи). Реальное время расчетов варьируется от нескольких суток до нескольких месяцев, а суммарное процессорное время может легко достигать сотен тысяч часов.
Программы визуализации - это приложения для десктопа, которые отображают трехмерные структуры молекул в разнообразных представлениях, позволяют анализировать их, рассчитывать различные параметры, а также создавать изображения и видеоролики высокого качества для презентаций и статей. В этой части статьи я опишу некоторые популярные вычислительные пакеты.


Gromacs распространяется по лицензии GPL, разрабатывается в системе Linux и по умолчанию использует открытые библиотеки и инструменты - библиотеку для преобразований Фурье FFTW3, компилятор GCC, систему сборки CMake, библиотеки BLAS, LAPACK, OpenMPI и т.п. В то же время, при наличии на кластере проприетарных библиотек и компиляторов (например, Intel MKL и ICC), их можно использовать для «выжимания» максимальной производительности.
Gromacs славится своей великолепной документацией. Руководство пользователя Gromacs - эталон качественной документации к научному ПО. Описаны базовые научные концепции, используемые алгоритмы и детали их реализации. Дотошно документированы форматы файлов данных, синтаксис и функциональность всех программ пакета, нюансы и сценарии их использования. Wiki-раздел официального сайта содержит огромное количество хорошо структурированной дополнительной информации, обучающих материалов, примеров. На худой конец можно воспользоваться списками рассылки, где вполне реально достучаться до разработчиков или опытных пользователей.

Иногда хочется занять свой ноутбук чем-нибудь полезным, да и так чтобы с работой помогало. Несколько лет назад, в университете сталкивался бегло с моделирование методом молекулярной динамики в gromacs, это хоть и не совсем то, но уже можно было притянуть за уши к моей работе. Русскоязычной информации по запуску моделирования в gromacs удалось найти крайне мало, примерно столько же и в глобальном масштабе, все в основном обсуждают уж очень глубокие/тонкие моменты. В итоге решил разобраться в нем, а заодно и написать краткое руководство, по использованию gromacs.
Преследуемая цель статьи — популяризировать Gromacs в широких кругах. Цели сделать полный охват метода и возможностей пакета нету, потому это будет краткий экскурс плохая методичка, с установками и запуском.
Немного слов… или зачем нам все это надо
Если говорить о моделировании атомных или молекулярных структур, то практически все методы можно разделить на две группы: Квантово-механические методы и методы молекулярной динамики. Отличие этих групп принципиальное.
Квантово-механические методы основываются на использовании квантовой механики. Используемые в них расчеты ведутся либо из “честных/точных” квантовых уравнений, либо из их некоторых приближений, которые делаются для ускорения расчетов. Стоит отметить, что данные методы могут считать химические реакции т.е. превращения одних веществ в другие с достаточно высокой точностью. Под точностью следует понимать значения энергий связей в системе, разницы до и после реакции. Для лучшего представления точности метода знаю, что бывали ситуации когда посчитанное значение оказывалось точнее измеренного экспериментального. Т.е. более точные эксперименты в будущем подтверждали квантово-механические расчеты. Но данные методы имеют существенный недостаток это требование к большой вычислительной мощности(на “не супер компьютерах” системы в тысячу атомов имеют не приличные времена расчетов). Какие-то цифры по системам я привести не могу, подзабыл.
Молекулярная-динамика основывается на классической ньютоновской механике. Т.е. все взаимодействия считаются классическим образом из “простых” уравнений. В этом и слабость и сила метода. С одной стороны это дает возможности моделировать системы с миллионами атомов. С другой стороны расчеты идут только на уровне межмолекулярного взаимодействия и расчет хим реакций «не возможны», в отличии от квантовой химии, где считается «всё».
Примеры использования gromacs:
Начнем с установки, т.к. она в принципе может вызвать трудности
Установка
Установка gromacs
Можно поставить какую-то сборку из репозитория:
но для хорошей производительности рекомендую произвести сборку самостоятельно, под свое железо(у меня это i5-3210M, geforce 620m):
Скачиваем последнюю, стабильную версию программы, в моем случае это gromacs 5.0.4. Распаковываем и создаем папку для сборки:
далее самый важный момент, из-за которого весь этот неприглядный процесс и затеян — выбор используемых комманд процессора, методов расчета, использования видеокарты итп. Подробнее о используемых методах указано в документации к установке. По порядку, используем для настройки cmake:
- использование FFTW: -DGMX_BUILD_OWN_FFTW=ON -DGMX_FFT_LIBRARY=FFTW
- выбираем набор процессорных команд, для моего процессора это avx и соответствующая запись -GMX_SIMD=AVX_256 (другие)
- поддержка видеокарты -DGMX_GPU=ON -DCUDA_TOOLKIT_ROOT_DIR=/usr/lib/nvidia-cuda-toolkit/ путь до cuda toolkit
- -DCMAKE_INSTALL_PREFIX=/usr/local/ выбираем папку установки. Если посмотреть на значения по-умолчанию, то можно увидеть, что установка произведется в /usr/local/gromacs/, что потребует дополнительного прописывания путей, для запуска, потому просто отправляем его в /usr/local.
Если на каком-то шаге начинает ругаться об отсутствующих пакетах — ставим их. У меня ни на что не срыгнулась, хотя ставил на свежую ubuntu 14.04, так что все должно пройти «гладко».
Программа ругается на отсутствие входных файлов, что и ожидалось.
Подробнее о установке можно найти или не найти в документации.
Установка VMD
VMD это программа визуализации молекул, т.е. просмотра результатов моделирования. Итак регистрируемся на сайте и скачиваем нужную нам версию VMD, рекомендую с поддержкой GPU.
Распаковываем, меняем если нужно пути установки и ставим, не забывая поменять название пакета на vmd и указать версию, при использовании checkinstall
Будем считать что этого нам достаточно и приступим к моделированию.
Моделирование
Моделирование можно разбить на несколько этапов.
- Решить что будет моделироваться и с какой целью, отсюда будут отталкиваться параметры моделирования.
- Найти/сделать файл с координатами молекул/ы (gro pdb)
- Создание топологии молекулы — т.е. сделать описание связей итп. Фактически сказать пакету gromacs с чем он будет работать
- Запуск моделирования
- релаксация
- само моделирование
Для наглядности попробуем смоделировать очень простую систему — воду. Координатный файл и файл топологии молекулы воды есть в силовых полях, которые вшиты в сам пакет. Т.к. моделирование чего-либо в воде производится достаточно часто, то существует несколько типов моделей воды и одна из лучших это tip4p-ew. Эту модель воды мы и будем использовать.
В папке громакса, уже есть отрелаксированная модель в 216 молекул воды, её и будем использовать.
Была скопирована отрелаксированная вода, в 216 молекул, далее она была размножена в трех направлениях «на еще одну», получился итоговый файл в 1728 молекул воды. Метод genconf является методом громакса, его описание можно найти в документации.Наглядно посмотрим за тем, что было сделано:
открывшаяся картинка, не очень не очень красива, сделаем по лучше, для этого откроем graphics->representation и выберем в «drawing method»: VDW![]()
теперь посмотрим, на большую картину «большой» файл воды, отжав D в отображении прошлого и используя file->new molecule, и так же меняем тип отрисовки атомов:
![]()
Теперь её отрелаксируем(т.к. 8 блоков на стыках скорее всего не отрелаксированны): для этого потребуются файлы параметра моделирования и топология молекулы, т.е. файлы отражающие то, какие связи в молекуле существуют, какие силы и на что могут действовать итп. Использовать будем поле сил OPLS/AA.
Для избегания возникновения граничных эффектов используется периодические боксы, т.е. на граниах система сталкивается сама с собой. Т.к. моделирование производится в какой-то среде, у которой есть температура, давление, то существуют методы привода к указанным параметрам. Это так называемые баростаты и термостаты, они бывают разных видов и используются на разных этапах моделирования.
Кулоновские сил распространяются на бесконечность, но при этом быстро спадают, потом используется так называемый радиус обрезания или верлет(подробнее в документации).
Для расчетов существует несколько разных алгоритмов, которые отличаются скоростью, и параметрами работы. Допустим некоторые алгоритмы не могут работать не сильно «дестабилизированных» системах, но при этом очень быстрые, когда система в некотором смысле стабильна. Другие же алгоритмы невероятно медлительны, но могут вывести систему из сильного отклонения в слабое итп.
Поля сил — это набор эмпирических параметров описывающих внутри и межмолекулярные взаимодействия, по сути это числа в лернард-джонсовское взимодействие(или морзе или. ), а так же параметры связи в молекуле. Т.е. сила стягивания связи, её длина, сила удержания плоских и двугранных углов итп. Все эти вещи достаточно подробно описаны в документации, а так же легко найти их описания на русском.
Разработчики рекомендуют, если не известно что нужно — использовать поле opls/aa.
Cоздаем файл описывающий топологию используемой системы:
Создаем папку для релаксации equi, в ней файл параметров моделирования следующего содержания:
Подробнее о параметрах моделирования можно почитать в документации. Количество используемых параметров может быть намного больше или меньше, зависит от требования условий. Используемые параметры, я
нечестно скопировал из примеров когда-то изучаемых мной курсов и для образовательных целей этого вполне хватит.Документация к gromacs достаточно подробная и если кто-то решит этим плотно заниматься, документацию стоит досконально прочитать, чтобы понимать в каком случае какие методы использовать, допустим того же учета кулоновского взаимодействия.
Запускаем релаксацию, из папки equi:
Чтобы просмотреть, что получилось необходимо произвести «перенос» атомов, которые ушли по граничным условиям в координатном файле моделирования, используя встроенную утилиту громакса trjconv. После чего открываем начальный файл координат vmd и подгружаем в него координаты:
В vmd нажимаем правой кнопкой на h2o_1728.gro, выбираем load data into molecule и там уже выбираем файл trajout.xtc и жмем load. Если кому-то интересно что будет без trjconv, то может подгрузить исходный traj_comp.xtc.Теперь используя прокрутку в vmd можно наблюдать за движением молекул в процессе моделирования.
По идеи для получения более-менее реальной модели необходимо произвести дальнейшее моделирование, поменяв термостат и баростат и увеличив время самого моделирования примерно в 5-10 раз. Но на воду саму по себе смотреть не очень интересно, куда интереснее будет поместит туда какое-то вещество, допустим сахар. Эта задача оказывается не только интереснее в плане наблюдения, но и в реализации, потому как топологию сахара придется создавать самим, а не брать готовую.
Сахар
Сахаров на самом деле всяких много, т.к. взять конкретный сахар цели нету, то берем первый понравившийся. Да и я особо уже не помню, чем они там по формулам различаются, фруктозы, сахарозы… Я нагуглил вот такой. Беру первый, как я понял это сахароза, посмотрим на него, уже стандартно в vmd, вот только отображение поставим CPK:
![]()
Теперь надо получить топологию данной молекулы, т.е. описать кто с кем связан и какие атомы соответствуют атомам в поле сил. Это можно сделать по-разному, регулярный способ мне не известен. Попробуем получить топологию из имеющихся уже данных — связи атомов в файле pdb и информации в поле силы для атомов.
В «хорошем» pdb файле уже описаны связи атомов, потому есть надежда что из него можно сконвертировать топлогию. И эта задача уже решена ранее, если порыться на сайте gromacs, то можно найти скрипты от участников, там и находится данная перловая утилита — topolgen-1.1.
используя её генерируем файл топологии:
если посмотреть на файл топологии то можно увидеть, что на позиции type стоят просто числа, а должны быть названия типов атомов из того набора сил, который используется. Для того чтобы соотнести данную молекулу с полем сил, надо посмотреть какие типы атомов используются, сколько у них связей и что им соответствует в наборе OPLS/AA.Для этого откроем в vmd снова молекулу, перейдем в representation, сверху нажмем «Create Rep» и сменим отображение на CPK, затем выберем вкладку selections. В Selected atoms удалим все имеющееся и введем serial 1, нажмем apply. Если посмотреть на отображение, то теперь один атом стал круглым, это и есть атом 1. Теперь мы можем соотнести атом с теми что имеются в наборе поля сил. Чтобы это сделать открываем файл поля сил atomtypes.atp, находящийся в /usr/local/share/gromacs/top/oplsaa.ff. И смотрим что подходит для атома углерода связанного с углеродом, кислородом и водородом, т.е. углерод в оксане. Такого там нету, потому будем просто искать подходящий по связям т.е. opls_193, далее меняем в vmd в selected atoms на 2 и ищем для второго атома, я выбрал opls_182 и так далее получаем в итоге файл топологии.
Так же включил поле сил, добавил название молекулы и сделал название основания.
приведена только измененная часть:Конвертируем координатный файл из pdb в gro, заодно помещая его в коробку 4 на 4 на 4:
Приводим его в соответствие с топологией, подписывая атомы и ставя название основания:Теперь пробуем запустить моделирование, для упрощения и проверки возьмем тот же файл grompp.mdp, что и использовали для воды.
Квантомеханические расчеты производятся в различных программах
гауссиан, и их описание заслуживает отдельных статей и вообще их использование трудозатратно. Но выходом из положения является некоторый сервис — ATB. Он позволяет удаленно на облаке рассчитывать не большие молекулы, а так же индексирует уже посчитанные, что позволяет найти необходимую топологию, сразу без расчетов. Однако используемое в нем поле сил gromos54a7, а значит соответствия атомов с полем сил opls/aa не будет(колонка type в top файлах). Выходом из ситуации может быть, как использования того же поля что и в ATB, так и модификация уже созданной нами топологии для задания внутримолекулярного взаимодействия, на основании расчетов в ATB, однако это требует экспериментальной проверки, т.к. «комбинация» из двух полей не лучший вариант.После не долгих поисков в репозитории ATB нашел сахарозу. Создадим папку ATB, куда и скачаем файлы топологии и расположения атомов(берем all-atoms). Получается ранее проделанные действия с сахаром были бесполезны, но целью их было показать построение топологии.
В итоге получили 2 файла sug.top и sug.pdb. Запустим моделирование на их основе и файла параметров моделирования, который использовали для воды – в файл топологии дописываем:добавлено в начало
Поместим 4 молекулы в одну «коробку»:
Возьмем файл параметров моделирования воды, увеличим время в 10 раз и запустим:
Результаты можно посмотреть в vmd.
Вода + сахар
Вода для данного силового поля рекомендованная spc, она хуже чем tip4p, но не будем усложнять и так перегруженную статью, используем spc.
Совместим воду и сахар:
Создаем файл топологии для смешанной системы:Далее запускаем моделирование:
Скорее всего получим ошибку, вызванную тем, что система сильно далеко от равновесия. Для решения данной проблемы — уменьшим шаг моделирования в 2 раза, до 0,001ps (в файле grompp.mdp строчка dt = 0.002; 2 fs). Для наглядности, я промоделировал до времени в 300ps. Результат приведен ниже.Для дальнейшего анализа можно использовать методы встроенные в gromacs такие как функции радиального распределения, энергии, температуры итп, подробнее можно найти в документации. На анализе я останавливаться не буду ибо статья и так уже по моим меркам большая.
Читайте также:


