
Синдром драйвер что это
Обновлено: 05.07.2024
Впервые синдром Драве был описан Charlotte Dravet как тяжелая миоклоническая эпилепсия младенцев. Хотя синдром часто включает в себя миоклонические феномены, они редко бывают первым проявлением заболевания. Полученные в ходе развития современных методов молекулярной генетики данные подтвердили основанные на клинических наблюдениях предположения (Veggiotti et al., 2001) о том, что синдром Драве является частью спектра тяжелых эпилепсий, клиническая картина которых может дополняться генерализованными тонико-клоническими припадками, фокальными припадками, атипичными абсансами и миоклониями.
Синдром Драве считается наиболее тяжелым расстройством спектра эпилептических синдромов, описываемых как «GEFS+» (Generalized Epilepsy with Febrile Seizures plus — генерализованная эпилепсия с фебрильными припадками плюс).
Более 80% случаев синдрома Драве вызвано вновь возникшей мутацией гена натриевого канала SCN1A (Claes et al., 2001). Описано более 100 новых мутаций (Mulley et al., 2005), наиболее часто «усеченные» мутации, но также сплайс-сайт мутации, делеции и миссенс-мутации. В оставшихся 20% случаев мутации гена SCNA1 отсутствуют. Сообщалось о двух семейных случаях мутаций гена GABRG2. Недавно Depienne et al. (2006) сообщили о соматическом и половом клеточном мозаицизме у родителей — бессимптомных носителей генов в двух семьях с двумя и более случаями синдрома Драве.
Начало, обычно в возрасте 4-10 месяцев, характеризуется клоническими припадками, часто односторонними и длительными, в 75% случаев возникающими при высокой температуре. Такие припадки неоднократно рецидивируют через короткие интервалы (обычно менее двух месяцев). В начале заболевания ребенок развивается нормально. В течение второго или третьего года жизни возникают другие типы припадков, включая парциальные припадки, атипичные абсансы, миоклонические судороги и эпизоды неконвульсивного статуса. В этот же период становится заметной задержка когнитивного развития.
Часто выявляются симптомы поражения пирамидного тракта и атаксия. Несмотря на часто повторяющиеся припадки, в течение первых месяцев заболевания на интериктальной ЭЭГ обычно не обнаруживается патологических изменений. Начиная со второго года жизни, на ЭЭГ отсутствуют медленные комплексы спайк-волна, появляются быстрые комплексы спайк-волна, часто вместе с мультифокальными пиками. Фоточувствительность имеется у 25% пациентов, и нередко наблюдается самопроизвольное начало припадка. Долгосрочный прогноз заболевания неблагоприятен. Сохраняются припадки в основном в виде тонико-клонических атак, а миоклонические припадки постепенно исчезают.
Во всех случаях присутствует умственная отсталость различной степени (Oguni et al., 2001b; Dravet и Bureau, 2005).

У некоторых пациентов генерализованные или односторонние клонические припадки, вызываемые небольшим подъемом температуры тела или даже горячей ванной, являются преобладающим или единственным видом припадков, тогда как короткие припадки могут не быть ведущим симптомом и могут не включать в себя миоклонические атаки, которые в любом случае редко являются выраженными проявлениями эпилепсии. Некоторые исследователи описывали подобные случаи как вариант миоклонической эпилепсии (Ogino et al., 1989; Watanabe et al., 1989), эпилепсии c геми-grand mal и медленными волнами с большой амплитудой на ЭЭГ (Doose, 1992) или как идиопатическую эпилепсию младенцев (Ebach et al., 2005).
Такие формы сгруппированы (Oguni et al., 2001а; Fukuma et al., 2004) в «пограничные случаи» тяжелой миоклонической эпилепсии. Течение при этом не отличается от типичных форм, хотя может быть менее тяжелым. Было показано, что некоторые из этих атипичных форм вызваны мутациями гена SCNA1, но часто это другие мутации, нежели те, которые вызывают типичные формы, особенно редко встречаются «усеченные» мутации (Claes et al., 2001; Fujivara et al., 2003; Fukumura et al., 2004). Тот же ген может быть ответственным за развитие и других эпилептических синдромов, включая эпилепсию с миоклонически-ми астатическими припадками и некоторые семейные случаи GEFS+ (Ebah et al, 2005).
Harkin et al. (2007) проанализировали 90 случаев «эпилептической энцефалопатии» с мутациями SCN1A: у 52 пациентов был типичный синдром Драве, у 25 — атипичные формы, у 6 была криптогенная генерализованная эпилепсия, у 4 — криптогенная фокальная эпилепсия, у 2 — миоклоническая-астатическая эпилепсия и у 1 —синдром Леннокса-Гасто, что демонстрирует варианты экспрессии гена.
Berkovic et al. (2006) выявили мутации SCN1A у 11 из 14 пациентов с так называемой вакцинальной энцефалопатией. Клинико-молекулярный корреляционный анализ выявил мутации в 8 случаях с фенотипом синдрома Драве, в 3 из 4 случаев пограничного синдрома Драве, и ни в одном из двух случаев синдрома Леннокса-Гасто. Эти данные, хотя и нуждаются в проверке, обеспечивают солидное альтернативное объяснение причинам возникновения подобных детских расстройств; эти случаи могут быть вызваны совершенно другими причинами, вне связи с вакцинацией, вопреки распространенному мнению.
Необходимо тщательно взвешивать взаимодействие стирипенто-ла с большим количеством других препаратов. При том, что полный контроль достигается в редких случаях, все же представляется, что эти комбинации уменьшают как частоту, так и длительность конвульсивных эпизодов. В прошлом вигабатрин давал хорошие результаты у пациентов с ослабленным миоклоническим компонентом. Наконец, применение топирамата дало обнадеживающие результаты в открытых исследованиях, вероятно, препарат должен назначаться в раннем периоде заболевания (Dravet и Bureau, 2005). Комбинация топирамата и стирипентола представляется простой и безопасной (Kroll-Seger et al„ 2006). Сообщалось, что ламотриджин вызывает ухудшение у маленьких детей с синдромом Драве (Guerrini et al, 1998b).
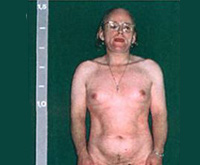
Адреногенитальный синдром — наследственное заболевание надпочечников, при котором вследствие функциональной несостоятельности ферментов нарушается стероидогенез. Проявляется вирилизацией гениталий, маскулиноподобным телосложением, недоразвитием груди, гирсутизмом, акне, аменореей или олигоменореей, бесплодием. В ходе диагностики определяют уровни 17-гидроксипрогестерона, 17-кетостероидов, андростендиона, АКТГ, проводят УЗИ яичников. Пациенткам назначают заместительную гормонотерапию глюкокортикоидами и минералокортикоидами, эстрогены в комбинации с андрогенами или прогестинами нового поколения. При необходимости выполняют пластику половых органов.
МКБ-10
Общие сведения
Адреногенитальный синдром, или врожденная дисфункция (гиперплазия) коры надпочечников, — наиболее частое из наследуемых заболеваний. Распространенность патологии отличается у представителей разных национальностей. Классические варианты АГС у лиц европеоидной расы встречаются с частотой 1:14 000 младенцев, в то время как у эскимосов Аляски этот показатель составляет 1:282.
Существенно выше заболеваемость у евреев. Так, неклассическую форму адреногенитального расстройства выявляют у 19% лиц еврейской национальности группы ашкенази. Патология передается по аутосомно-рецессивному типу. Вероятность рождения ребенка с таким синдромом при носительстве патологического гена у обоих родителей достигает 25%, в браке носителя и больного — 75%. Если один из родителей имеет полноценные ДНК, клинические проявления синдрома у детей не развиваются. При наличии АДС у отца и матери ребенок также будет болен.
Причины
У больных с наследуемой гиперплазией надпочечников генетический дефект проявляется несостоятельностью ферментных систем, участвующих в секреции стероидных гормонов. В 90-95% случаев патология возникает при повреждении гена, который отвечает за синтез 21-гидроксилазы — фермента, влияющего на образование кортизола. В остальных клинических случаях вследствие дефекта ДНК нарушается производство других ферментов, обеспечивающих стероидогенез, — StAR/20,22-десмолазы, 3-β-гидрокси-стероиддегидрогеназы, 17-α-гидроксилазы/17,20-лиазы, 11-β-гидроксилазы, P450-оксидоредуктазы и синтетазы альдостерона.
У пациентов с признаками вирилизирующего синдрома вместо активного гена CYP21-B в коротком плече 6-й аутосомы расположен функционально несостоятельный псевдоген CYP21-A. Структура этих участков ДНК-цепи во многом гомологична, что повышает вероятность конверсии генов в мейозе с перемещением участка нормального гена на псевдоген или делецию CYP21-B.
По-видимому, именно этими механизмами объясняется существование скрытых форм болезни, дебютирующих в пубертате или постпубертатном периоде. В таких случаях клинические признаки патологии становятся заметными после нагрузок, истощающих кору надпочечников: тяжелых болезней, травм, отравлений, радиационных воздействий, длительного периода интенсивной работы, психологически напряженных ситуаций и т. д.
Патогенез
В основе механизма развития наиболее распространенного варианта адреногенитального синдрома с дефектом CYP21-B-гена лежит принцип обратной связи. Ее начальным звеном становится дефицит стероидов — кортизола и альдостерона. Несостоятельность процессов гидроксилирования сопровождается неполным переходом 17-гидроксипрогестерона и прогестерона в 11-дезоксикортизол и дезоксикортикостерон. В результате снижается секреция кортизола, а для компенсации этого процесса в гипофизе усиливается синтез АКТГ — гормона, вызывающего компенсаторную гиперплазию коры надпочечников для стимуляции выработки кортикостероидов.
Параллельно возрастает синтез андрогенов и появляются видимые признаки их влияния на чувствительные ткани и органы. При умеренном снижении активности фермента минералокортикоидная недостаточность не развивается, поскольку потребность организма в альдостероне почти в 200 раз ниже по сравнению с кортизолом. Только глубокий дефект гена вызывает тяжелую клиническую симптоматику, которая проявляется с раннего возраста. Патогенез развития заболевания при нарушении структуры других участков ДНК аналогичен, однако пусковым моментом являются нарушения в других звеньях стероидогенеза.
Классификация
Систематизация различных форм вирилизирующей гиперплазии надпочечников основана на особенностях клинической картины заболевания, выраженности генетического дефекта и времени проявления первых патологических признаков. Тяжесть расстройства напрямую связана со степенью повреждения ДНК. Специалисты в сфере эндокринологии различают следующие виды адреногенитального синдрома:
- Сольтеряющий. Самый тяжелый вариант патологии, проявляющийся в первый год жизни ребенка грубыми нарушениями строения наружных половых органов у девочек и их увеличением у мальчиков. Активность 21-гидроксилазы составляет не более 1%. Значительное нарушение стероидогенеза приводит к выраженным соматическим нарушениям — рвоте, поносу, судорогам, чрезмерной пигментации кожи. Без лечения такие дети умирают в раннем возрасте.
- Простой вирильный. Течение заболевания менее тяжелое, чем при сольтеряющем варианте. Преобладают проявления неправильного развития гениталий у младенцев женского пола, увеличение их размеров у мальчиков. Признаки надпочечниковой недостаточности отсутствуют. Уровень активности 21-гидроксилазы снижен до 1-5%. С возрастом у пациентов нарастают признаки вирилизации вследствие стимулирующего действия андрогенов.
- Неклассический (постпубертатный). Наиболее благоприятная форма АГС, явные признаки которой возникают в период полового созревания и в репродуктивном возрасте. Наружные половые органы имеют нормальное строение, может быть увеличен клитор у женщин и половой член у мужчин. Функциональность 21-гидроксилазы снижена до 20-30%. Заболевание выявляется случайно при обследовании в связи с бесплодием или нарушениями менструальной функции.
Сольтеряющий и простой вирильный виды адреногенитальных расстройств относят к категории антенатальной патологии, формирующейся внутриутробно и проявляющейся с момента рождения. При дефекте строения других генов наблюдаются более редкие варианты заболевания: гипертензивные — классический (врожденный) и неклассический (поздний), гипертермический, липидный, с ведущими проявлениями гирсутизма.
Симптомы
Сольтеряющий и простой вирильный
При антенатальных формах заболевания основным клиническим симптомом является видимая вирилизация гениталий. У новорожденных девочек обнаруживаются признаки женского псевдогермафродитизма. Клитор большой по размерам или имеет пенисообразную форму, преддверие влагалища углублено, сформирован урогенитальный синус, большие и малые половые губы увеличены, промежность высокая. Внутренние половые органы развиты нормально.
У младенцев-мальчиков увеличен половой член и гиперпигментирована мошонка. Кроме того, при сольтеряющем адреногенитальном расстройстве выражена симптоматика надпочечниковой недостаточности с тяжелыми, зачастую несовместимыми с жизнью соматическими нарушениями (понос, рвота, судороги, обезвоживание и др.), которые проявляются с 2-3-недельного возраста. У девочек с простым вирильным АГС по мере взросления признаки вирилизации усиливаются, формируется диспластическое телосложение.
Из-за ускорения процессов окостенения пациентки отличаются невысоким ростом, широкими плечами, узким тазом, короткими конечностями. Трубчатые кости массивные. Половое созревание начинается рано (до 7 лет) и протекает с развитием вторичных мужских половых признаков. Отмечается увеличение клитора, снижение тембра голоса, нарастание мышечной силы, формирование типичной для мужчин формы перстневидного хряща щитовидной железы. Грудь не растет, менархе отсутствует.
Неклассический
Менее специфичны клинические симптомы при неклассических формах вирилизирующего синдрома, возникшие в пубертате и после стрессовых нагрузок (выкидыша на ранних сроках беременности, медицинского аборта, операции и др.). Обычно пациентки вспоминают, что у них еще в младшем школьном возрасте появилось небольшое оволосение в подмышечных впадинах и на лобке. В последующем развились признаки гирсутизма с ростом стержневых волос над верхней губой, по белой линии живота, в области грудины, в сосково-ареолярной зоне.
Женщины с АГС предъявляют жалобы на стойкую угревую сыпь, пористость и повышенную жирность кожи. Менархе наступает поздно — к 15-16 годам. Менструальный цикл неустойчив, интервалы между менструациями достигают 35-45 дней и более. Кровянистые выделения во время месячных скудные. Молочные железы небольшие. Клитор несколько увеличен. Такие девушки и женщины могут иметь высокий рост, узкий таз, широкие плечи.
По наблюдениям специалистов в сфере акушерства и гинекологии, чем позже развиваются адреногенитальные расстройства, тем менее заметны внешние признаки, характерные для мужчин, и тем чаще ведущим симптомом становится нарушение месячного цикла. При более редких генетических дефектах пациентки могут жаловаться на повышение артериального давления или, наоборот, гипотонию с низкой работоспособностью и частыми головными болями, гиперпигментацию кожи с минимальными симптомами вирилизации.
Осложнения
Основным осложнением адреногенитального синдрома, по поводу которого пациентки обращаются к акушерам-гинекологам, является стойкое бесплодие. Чем раньше проявилось заболевание, тем меньше вероятность забеременеть. При значительной ферментной недостаточности и клинических проявлениях простого вирилизирующего синдрома беременность вообще не наступает.
У забеременевших пациенток с пубертатными и постпубертатными формами заболевания возникают самопроизвольные выкидыши на раннем сроке. В родах возможна функциональная истмико-цервикальная недостаточность. Такие женщины более склонны к возникновению психоэмоциональных расстройств — склонности к депрессии, суицидальному поведению, проявлениям агрессии.
Диагностика
Постановка диагноза при антенатальных типах АГС с характерными изменениями половых органов не представляет сложности и проводится сразу после родов. В сомнительных случаях применяют кариотипирование для подтверждения женского кариотипа (46ХХ), молекулярно-генетические тесты. Большее значение диагностический поиск приобретает при позднем клиническом дебюте или скрытом течении с минимальными внешними проявлениями вирилизации. В подобных ситуациях для выявления адреногенитального синдрома используют следующие лабораторные и инструментальные методы:
- Уровень 17-ОН-прогестерона. Высокая концентрация 17-гидроксипрогестерона, который является предшественником кортизола — ключевой признак недостаточности 21-гидроксилазы. Его содержание увеличено в 3-9 раз (от 15 нмоль/л и выше).
- Стероидный профиль (17-КС). Повышение уровня 17-кетостероидов в моче у женщин в 6-8 раз свидетельствует о высоком содержании андрогенов, производимых корой надпочечников. При выполнении преднизолоновой пробы концентрация 17-КС уменьшается на 50-75%.
- Содержание андростендиона в сыворотке крови. Повышенные показатели этого высокоспецифичного метода лабораторной диагностики подтверждают усиленную секрецию предшественников мужских половых гормонов.
- Уровень АКТГ в крови. Для классических форм заболевания характерна компенсаторная гиперсекреция адренокортикотропного гормона передней долей гипофиза. Поэтому при синдроме вирилизирующей дисфункции показатель повышен.
- УЗИ яичников. В корковом веществе определяются фолликулы на разных стадиях созревания, не достигающие преовуляторных размеров. Яичники могут быть несколько увеличены, однако разрастания стромы не наблюдается.
- Измерение базальной температуры. Температурная кривая типична для ановуляторного цикла: первая фаза растянута, вторая укорочена, что обусловлено недостаточностью желтого тела, которое не образуется из-за отсутствия овуляции.
Для сольтеряющего варианта АГС также характерна повышенная концентрация ренина в плазме крови. Дифференциальная диагностика адреногенитальных расстройств, возникших в пубертатном и детородном возрасте, проводится с синдромом поликистозных яичников, овариальными андробластомами, андростеромами надпочечников, вирильным синдромом гипоталамического происхождения и конституциональным гирсутизмом. В сложных случаях к диагностике привлекают эндокринологов, урологов, врачей-генетиков.
Лечение адреногенитального синдрома
Основным способом коррекции вирильной дисфункции надпочечников является заместительная гормональная терапия, восполняющая дефицит глюкокортикоидов. Если у женщины со скрытым АГС нет репродуктивных планов, кожные проявления гиперандрогении незначительны и месячные ритмичны, гормоны не применяют. В остальных случаях выбор схемы лечения зависит от формы эндокринной патологии, ведущей симптоматики и степени ее выраженности. Зачастую назначение глюкокортикоидных препаратов дополняют другими медикаментозными и хирургическими методами, подобранными в соответствии с конкретной терапевтической целью:
- Лечение бесплодия. При наличии планов по деторождению женщина под контролем андрогенов крови принимает глюкокортикоиды до полного восстановления овуляторного месячного цикла и наступления беременности. В резистентных случаях дополнительно назначают стимуляторы овуляции. Во избежание выкидыша гормонотерапию продолжают до 13-й недели гестационного срока. В I триместре также рекомендованы эстрогены, во II-III — аналоги прогестерона, не обладающие андрогенным эффектом.
- Коррекция нерегулярных месячных и вирилизации. Если пациентка не планирует беременность, но жалуется на расстройство менструального цикла, гирсутизм, угри, предпочтительны средства с эстрогенным и антиандрогенным эффектом, оральные контрацептивы, содержащие гестагены последнего поколения. Терапевтический эффект достигается за 3-6 месяцев, однако по окончании лечения при отсутствии заместительной гормонотерапии признаки гиперандрогении восстанавливаются.
- Лечение врожденных форм АГС. Девочкам с признаками ложного гермафродитизма проводят адекватную гормонотерапию и выполняют хирургическую коррекцию формы половых органов — клитеротомию, интроитопластику (вскрытие урогенитального синуса). При сольтеряющих адреногенитальных расстройствах кроме глюкокортикоидов под контролем рениновой активности назначают минералокортикоиды с увеличением терапевтических доз при возникновении интеркуррентных заболеваний.
Определенные сложности в ведении пациентки возникают в тех случаях, когда заболевание не диагностировано в акушерском стационаре, и девочка с выраженной вирилизацией гениталий регистрируется и воспитывается как мальчик. При решении о восстановлении женской половой идентичности хирургическую пластику и гормонотерапию дополняют психотерапевтической поддержкой. Решение о сохранении гражданского мужского пола и удалении матки с придатками принимается в исключительных случаях по настоянию больных, однако такой подход считается ошибочным.
Прогноз и профилактика
Прогноз при своевременном обнаружении адреногенитального синдрома и адекватно подобранной терапии благоприятный. Даже у пациенток со значительной вирилизацией гениталий после пластической операции возможна нормальная половая жизнь и естественные роды. Заместительная гормонотерапия при любой форме АГС способствует быстрой феминизации — развитию грудных желез, появлению месячных, нормализации овариального цикла, восстановлению генеративной функции. Профилактика заболевания осуществляется на этапе планирования беременности.
Если в роду наблюдались случаи подобной патологии, показана консультация генетика. Проведение пробы с АКТГ обоим супругам позволяет диагностировать гетерозиготное носительство или скрытые формы адреногенитального расстройства. При беременности синдром может быть обнаружен по результатам генетического анализа клеток хорионических ворсин или содержимого околоплодных вод, полученных методом амниоцентеза. Неонатальный скрининг, проводимый на 5-е сутки после родов, направлен на выявление повышенной концентрации 17-гидропрогестерона для быстрого выбора терапевтической тактики.
Синдром Драве – это детская энцефалопатия наследственного характера, которая характеризуется эпилептиформными приступами, отставанием в психическом развитии и резистентностью к противоэпилептической терапии. Клинически заболевание проявляется полиморфными эпилептическими припадками, неврологическими расстройствами, атипическими абсансами и фокальными моторными пароксизмами. Диагностика синдрома Драве базируется на характеристике возникающих приступов, данных ЭЭГ и МРТ, идентификации мутации генов SCN1A или GABRG2. Лечение малоэффективно и проводится с целью уменьшения частоты приступов, профилактики эпилептического статуса.
Общие сведения
Синдром Драве или тяжелая миоклоническая эпилепсия младенчества – это аутосомно-доминантная энцефалопатия с дебютом в первые 12 месяцев жизни ребенка, которая проявляется фебрильными и афебрильными генерализованными приступами, фокальными миоклоническими пароксизмами, расстройствами неврологического статуса и дефицитом интеллекта. Впервые заболевание было описано французским психиатром и эпилептологом Шарлоттой Драве в 1978 году. Встречается данный синдром редко, распространенность – 1:20-40 тысяч детского населения. У мальчиков патология возникает вдвое чаще, чем у девочек. Исход синдрома Драве неблагоприятный – заболевание неизлечимо и слабо поддается медикаментозной терапии. Летальность составляет порядка 16-18%.
Причины и симптомы синдрома Драве
Синдром Драве – это генетически детерминированная патология, которая передается по аутосомно-доминантному типу наследования. Спровоцировать развитие тяжелой миоклонической эпилепсии младенчества могут мутации локуса SCN1A на 24 участке длинного плеча 2 хромосомы (в 80% случаев) или GABRG2 на 5q34. Данные гены кодируют α1-субъединицу Na+-каналов, что приводит к нарушению физиологических процессов реполяризации и деполяризации в нейронах, и как следствие – к патологической активности ЦНС.
В клинической картине синдрома Драве выделяют 3 этапа развития: фебрильный (до 12-24 месяцев), агрессивный или катастрофический (2-8 лет), статический (старше 8 лет). Дебют заболевания происходит в возрасте от 2 месяцев до 1 года, в среднем – в 5 месяцев. До момента возникновения первых симптомов ребенок развивается нормально, неврологических и психических отклонений не наблюдается. В большинстве случаев первичными проявлениями фебрильной стадии синдрома Драве становятся фибриллярные судороги атипического характера. Они имеют большую продолжительность (свыше 20 минут), включают в себя очаговые компоненты и альтернирующие гемиконвульсии, иногда переходят в эпилептический припадок. На ранних этапах такие состояния сопровождаются субфебрильной или фебрильной температурой тела, в дальнейшем подобных проявлений не наблюдается. Зачастую при синдроме Драве приступ может быть спровоцирован гипертермией (согреванием, горячей ванной или инфекционной патологией), световыми раздражителями, резкими движениями и т. д.
Катастрофический или агрессивный период синдрома Драве характеризуется выраженными полиморфными клонико-тонико-клоническими припадками, альтернирующими гемиконвульсиями, очаговыми моторными пароксизмами, атипичными абсансами. Приступы начинаются с мышечных подергиваний по всему телу (иногда – асинхронных), переходят в кратковременную тоническую, а затем – клоническую фазы. Часто подобное состояние трансформируется в эпилептический статус, который может сохраняться до нескольких суток. В возрасте 1-2 лет у больных с синдромом Драве определяется дефицит интеллекта (олигофрения) и гиперактивность, поведенческие аномалии, нарастающие до 6-7 лет и сохраняющиеся на протяжении всей жизни. Также развиваются неврологические нарушения: мышечная гипотония, атаксия, интенционный тремор, моторная неловкость, признаки пирамидной недостаточности. В этом же возрасте у части детей возникает паттерн-сенситивность, при которой определенная одежда, обои или телевизионные передачи могут стать причиной очередного приступа.
Статическая стадия синдрома Драве характеризуется уменьшением интенсивности и частоты эпилептических припадков. Психические и неврологические отклонения остаются. Большая часть приступов возникает в ночное время или сразу после пробуждения. Как и в других периодах, они могут быть спровоцированы повышением температуры тела, ярким светом, резким движением и др. На фоне отставания в интеллектуальном развитии, нарушений психики и резистентности заболевания к лечению пациент почти полностью лишен способности адаптироваться в социуме.
Диагностика синдрома Драве
Диагностика синдрома Драве основывается на анамнестических данных, физикальном обследовании, лабораторных и инструментальных методах исследования. Из анамнеза педиатром выясняется возраст, в котором произошла манифестация патологии, первичные проявления, характеристика приступов, степень их тяжести и динамика развития. При осмотре ребенка в межприступный период можно выявить отставание в интеллектуальном развитии (ЗПР), гиперактивность, нарушения неврологического статуса. Во время припадка определяются атипичные абсансы, очаговые расстройства, альтернирующие гемиконвульсии.
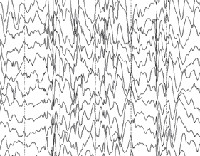
Общие лабораторные анализы (ОАК, ОАМ, анализ кала) малоинформативны – выраженные отклонения от возрастной нормы, как правило, отсутствуют. Из инструментальных методов исследования при синдроме Драве используются электроэнцефалограмма (ЭЭГ) и магнитно-резонансная томография (МРТ). Между приступами на ЭЭГ у большинства таких детей определяется сочетание очаговой, мультирегиональной и диффузной эпилептиформной активности с нарастанием во сне. При низкой частоте припадков данные признаки могут отсутствовать. По результатам МРТ головного мозга удается установить признаки диффузной атрофии коры головного мозга и мозжечка, субкортикальных слоев, иногда – увеличение размеров желудочков. Для подтверждения синдрома Драве используется кариотипирование с определением мутации генов SCN1A или GABRG2.
В педиатрии дифференциальная диагностика синдрома Драве проводится с фебрильными судорогами, митохондриальными и дисметаболическими патологиями, доброкачественной миоклонической эпилепсией младенчества, синдромами Леннокса-Гасто и Дозе, другими формами эпилепсии у детей, которые сопровождаются миоклоническими припадками. Практически идентичную клиническую картину имеет мутация гена PCDH19 – эпилепсия с умственной отсталостью, ограниченная женским полом.
Лечение синдрома Драве
Синдром Драве – это форма эпилепсии у детей, которая почти не поддается терапии. Основная цель лечения – снизить чистоту приступов, профилактировать их трансформацию в эпилептический статус. Как правило, большинство распространенных противоэпилептических средств при тяжелой миоклонической эпилепсии младенчества неэффективны. В качестве стартовой терапии показаны вальпроаты (вальпроева кислота) и сульфат-замещенные моносахариды (топирамат). Также могут применяться фармакологические средства из групп барбитуратов и бензодиазепинов. В некоторых случаях при синдроме Драве позитивная динамика отмечается на фоне кетогенной диеты, которая подразумевает большое количество жиров и строгое ограничение углеводов.
Прогноз и профилактика синдрома Драве
Прогноз для жизни при синдроме Драве сомнительный, для выздоровления – неблагоприятный. Дефицит интеллекта, расстройства психики, эпилептические припадки и неврологические нарушения обычно сохраняются на протяжении всей жизни человека, что обусловливает его полную социальную дезадаптацию. Обычно приступы возникают в ночное время или сразу после пробуждения, а их интенсивность и частота уменьшаются. Смертность составляет порядка 15,9-18%. Основные причины – синдром внезапной детской смерти при эпилепсии, интеркуррентные инфекционные заболевания, несчастные случаи во время припадков.
Антенатальная профилактика синдрома Драве аналогична другим наследственным заболеваниям. Она подразумевает медико-генетическое консультирование и планирование беременности, кариотипирование плода посредством амнио- или кордоцентеза. Постнатальные превентивные меры включают в себя исключение гипертермических состояний у ребенка (раннее лечение инфекционных заболеваний, избегание горячих ванн и т. д.) и других факторов, которые могут спровоцировать приступ.

Синдром Драве - редкое заболевание, характеризующееся судорогами и проблемами в развитии. Приступы начинаются в возрасте до 1 года, когнитивные, поведенческие и физические проблемы - примерно в возрасте 2-3 лет.
Синдром Драве - это пожизненное состояние. Заболевание связано с генетическим дефектом в гене SCN1A, хотя оно может возникать и без генетического дефекта.
Припадки, вызванные синдромом Драве, особенно трудно контролировать. Противосудорожные препараты, которые обычно используются при большинстве судорожных расстройств, не эффективны, однако поиски новых методов терапии продолжаются.
Симптомы синдрома Драве
Судороги - самый ранний симптом синдрома Драве. Проблемы развития, а также судороги, как правило, ухудшаются по мере взросления ребенка.
Симптомы синдрома Драве включают в себя:
Судороги часто связаны с лихорадкой, хотя они могут возникать и без нее.
Существует несколько типов припадков, которые обычно возникают при синдроме Драве, включая миоклонические, тонико-клонические и не судорожные припадки. Для этого расстройства также характерны длительные припадки и эпилептический статус.
На самом деле, первый приступ может быть особенно длительным по продолжительности.
- Люди с синдромом Драве могут иметь светочувствительность, которая представляет собой тенденцию к припадкам в ответ на яркие или мигающие огни.
- Кроме того, человек с синдромом Драве может быть склонен к судорогам в ответ на изменения температуры тела.
Трудности с координацией и ходьбой, известные как атаксия, начинаются в детстве и продолжаются в подростковом и взрослом возрасте.
Люди, живущие с синдромом Драве, имеют согнутое положение при ходьбе. Часто присутствует низкий мышечный тонус, который может проявляться как мышечная слабость.
У детей могут развиться речевые и когнитивные проблемы, которые сохраняются на протяжении всей жизни. При синдроме Драве может быть целый ряд когнитивных способностей, и некоторые люди с этим заболеванием обладают более высокой способностью к обучению, чем другие.
Дети и взрослые, живущие с синдромом Драве, могут проявлять раздражительность, агрессию или поведение, напоминающее аутизм.
Люди с синдромом Драве склонны к инфекциям.
- Нарушения потоотделения и терморегуляции
Люди с синдромом Драве могут испытывать изменения в вегетативной нервной системе, особенно вызывающие снижение потоотделения и чрезмерно высокую или низкую температуру тела.
Синдром Драве связан с хрупкими костями и предрасположенностью к переломам костей.
Около трети людей, живущих с синдромом Драве, имеют нерегулярное сердцебиение, такое как учащенное сердцебиение, медленное сердцебиение или другое нарушение, такое как длительный интервал QT.
Прогноз развития заболевания
Синдром Драве - это пожизненное состояние, симптомы которого со временем улучшаются. Существует повышенный риск ранней смерти, часто связанный с травмами, вызванными судорогами.
Люди с синдромом Драве также более склонны к внезапной неожиданной смерти при эпилепсии (SUDEP) - состоянии, при котором происходит неожиданная смерть, обычно во время сна.
Причины синдрома Драве
Считается, что синдром Драве вызван дефектом функции натриевых каналов и описывается как форма каналопатии. Натриевые каналы регулируют работу мозга и нервов. Дефект функции натриевых каналов может вызвать целый ряд проблем, включая неустойчивую мозговую активность, проявляющуюся в виде судорог, и дефектную связь между клетками мозга, проявляющуюся в виде нарушений развития.
Генетика
Около 80% людей с синдромом Драве имеют дефект во второй хромосоме гена SCN1A, который кодирует натриевые каналы. Этот дефект возникает по наследственной схеме, мутация также может впервые возникнуть у пострадавшего ребенка.
Постановка диагноза
Синдром Драве диагностируется на основании клинической оценки врача.
По данным Фонда синдрома Драве, клинические характеристики синдрома Драве включают, по крайней мере, четыре из следующих пяти характеристик:
- Нормальное когнитивное и моторное развитие до первого приступа
- Два или более приступов в возрасте до 1 года
- Сочетание миоклонических, гемиклонических или генерализованных тонико-клонических припадков
- Два или более приступов, длящихся более 10 минут
- Отсутствие улучшения при стандартном противосудорожном лечении и продолжающиеся судороги после двух лет
Диагностические тесты
ЭЭГ обычно нормальна, когда у человека с синдромом Драве нет приступов, особенно у очень маленьких детей. ЭЭГ покажет отклонения, соответствующие активности припадка во время припадка. В более позднем детстве, а также в подростковом и взрослом возрасте ЭЭГ также может демонстрировать замедление между приступами.
Как правило, МРТ головного мозга человека с синдромом Драве, как ожидается, будет нормальной. Это может свидетельствовать об атрофии (истончении) гиппокампа или всего мозга в зрелом возрасте.
Генетическое тестирование может выявить мутацию SCN1A, которая чаще всего присутствует у людей с синдромом Драве. Это было отмечено в мозаичном узоре, что означает, что у человека могут быть некоторые клетки с мутацией, а некоторые без нее.
Лечение синдрома Драве
Существует целый ряд различных проблем, с которыми может столкнуться человек с синдромом Драве, и все они трудно поддаются лечению. Лечение физических, когнитивных и поведенческих проблем синдрома Драве индивидуализировано и может включать физиотерапию, логопедию и поведенческую терапию.
Припадки нелегко контролировать. Как правило, противосудорожные средства, используемые при синдроме Драве, включают комбинацию вальпроата, клобазама, стирипентола, топирамата, леветирацетама и бромидов.
Для лечения судорог также рассматриваются кетогенная диета и стимуляция блуждающего нерва.
В июне 2018 года Управление по контролю за продуктами и лекарствами США (FDA) одобрило Эпидиолекс (каннабидиол) для лечения синдрома Драве, а также другого синдрома эпилепсии - синдрома Леннокса Гасто. Более ранние исследования показали, что дети с синдромом Драве испытывали снижение частоты приступов при приеме Эпидиолекса и были способны переносить препарат.
Лекарства,ухудшающие синдром Драве
Стандартные противосудорожные средства, которые, как полагают, оказывают влияние на натриевые каналы, включают карбамазепин, окскарбазепин, фенитоин и ламотриджин. Это может ухудшить, а не улучшить судороги при синдроме Драве.
Читайте также: