
Как установить fastqc windows
Обновлено: 06.07.2024
FASTQ format is a text-based format for storing both a biological sequence (usually nucleotide sequence) and its corresponding quality scores. Both the sequence letter and quality score are each encoded with a single ASCII character for brevity.
It was originally developed at the Wellcome Trust Sanger Institute to bundle a FASTA formatted sequence and its quality data, but has recently become the de facto standard for storing the output of high-throughput sequencing instruments such as the Illumina Genome Analyzer. [1]
Contents
Format [ edit ]
A FASTQ file normally uses four lines per sequence.
A FASTQ file containing a single sequence might look like this:
The original Sanger FASTQ files also allowed the sequence and quality strings to be wrapped (split over multiple lines), but this is generally discouraged as it can make parsing complicated due to the unfortunate choice of "@" and "+" as markers (these characters can also occur in the quality string).
Illumina sequence identifiers [ edit ]
Sequences from the Illumina software use a systematic identifier:
Note that more recent versions of Illumina software output a sample number (as taken from the sample sheet) in place of an index sequence. For example, the following header might appear in the first sample of a batch:
NCBI Sequence Read Archive [ edit ]
FASTQ files from the INSDC Sequence Read Archive often include a description, e.g.
In this example there is an NCBI-assigned identifier, and the description holds the original identifier from Solexa/Illumina (as described above) plus the read length. Sequencing was performed in paired-end mode (
500bp insert size), see SRR001666. The default output format of fastq-dump produces entire spots, containing any technical reads and typically single or paired-end biological reads. Its behavior was driven by the demands of several projects current at the time fastq-dump was developed, most notably the 1000 Genomes Project.
Modern usage of FASTQ almost always involves splitting the spot into its biological reads, as described in submitter-provided metadata:
When present in the archive, fastq-dump can attempt to restore read names to original format. NCBI does not store original read names by default:
Also note that fastq-dump converts this FASTQ data from the original Solexa/Illumina encoding to the Sanger standard (see encodings below). This is because the SRA serves as a repository for NGS information, rather than format. The various *-dump tools are capable of producing data in several formats from the same source. The requirements for doing so have been dictated by users over several years, with the majority of early demand coming from the 1000 Genomes Project.
Variations [ edit ]
Quality [ edit ]
Q sanger = − 10 log 10 p <displaystyle Q_< ext >=-10,log _<10>p>
The Solexa pipeline (i.e., the software delivered with the Illumina Genome Analyzer) earlier used a different mapping, encoding the odds p/(1-p) instead of the probability p:
Q solexa-prior to v.1.3 = − 10 log 10 p 1 − p <displaystyle Q_< ext>=-10,log _<10><frac
Although both mappings are asymptotically identical at higher quality values, they differ at lower quality levels (i.e., approximately p > 0.05, or equivalently, Q [2] In retrospect, this entry in the manual appears to have been an error. The user gu > Q phred = − 10 log 10 e <displaystyle Q_< ext
>=-10log _< ext<10>>e> , where e is the estimated probability of a base being wrong. [3]
Encoding [ edit ]
- Starting in Illumina 1.8, the quality scores have basically returned to the use of the Sanger format (Phred+33).
Color space [ edit ]
Simulation [ edit ]
FASTQ read simulation has been approached by several tools. [9] [10] A comparison of those tools can be seen here. [11]
Compression [ edit ]
Quality values account for about half of the required disk space in the FASTQ format (before compression), and therefore the compression of the quality values can significantly reduce storage requirements and speed up analysis and transmission of sequencing data. Both lossless and lossy compression are recently being cons >[12] performs lossy compression with a rate (number of bits per quality value) specified by the user. Based on rate-distortion theory results, it allocates the number of bits so as to minimize the MSE (mean squared error) between the original (uncompressed) and the reconstructed (after compression) quality values. Other algorithms for compression of quality values include SCALCE [13] and Fastqz. [14] Both are lossless compression algorithms that prov >[15] .
As of the HiSeq 2500 Illumina gives the option to output qualities that have been coarse grained into quality bins. The binned scores are computed directly from the empirical quality score table, which is itself tied to the hardware, software and chemistry that were used during the sequencing experiment. [16]
Encryption [ edit ]
The encryption of FASTQ files has been mostly addressed with a specific encryption tool: Cryfa. [17] Cryfa uses AES encryption and enables to compact data besides encryption. It can also address FASTA files.
File extension [ edit ]
There is no standard file extension for a FASTQ file, but .fq and .fastq, are commonly used.
Format converters [ edit ]
- Biopython version 1.51 onwards (interconverts Sanger, Solexa and Illumina 1.3+)
- EMBOSS version 6.1.0 patch 1 onwards (interconverts Sanger, Solexa and Illumina 1.3+)
- BioPerl version 1.6.1 onwards (interconverts Sanger, Solexa and Illumina 1.3+)
- BioRuby version 1.4.0 onwards (interconverts Sanger, Solexa and Illumina 1.3+)
- BioJava version 1.7.1 onwards (interconverts Sanger, Solexa and Illumina 1.3+)
- MAQ can convert from Solexa to Sanger (use this patch to support Illumina 1.3+ files).
- fastx_toolkit The included fastq_quality_converter program can convert Illumina to Sanger
Command line conversions [ edit ]
FASTQ to FASTA format:
Illumina FASTQ 1.8 to 1.3
Illumina FASTQ 1.3 to 1.8
Illumina FASTQ 1.8 raw quality to binned quality (HiSeq Qtable 2.10.1, HiSeq 4000 )
Illumina FASTQ 1.8 raw quality to clinto format (a visual block representation)
1 расширения(ы) и 0 псевдоним(ы) в нашей базе данных
Ниже вы можете найти ответы на следующие вопросы:
- Что такое .fastq файл?
- Какая программа может создать .fastq файл?
- Где можно найти описание .fastq формат?
- Что может конвертировать .fastq файлы в другой формат?
- Какие MIME-тип связан с .fastq расширение?
FASTQ Sequence
Другие типы файлов могут также использовать .fastq расширение файла. Если у вас есть полезная информация о .fastq расширение, написать нам!
Не удается открыть .fastq файл?
Не удалось открыть этот файл:
Чтобы открыть этот файл, Windows необходимо знать, какую программу вы хотите использовать, чтобы открыть его. Окна могут выходить в интернет, чтобы искать его автоматически, или вы можете вручную выбрать из списка программ, установленных на вашем компьютере.
Чтобы изменить ассоциации файлов:
- Щелкните правой кнопкой мыши файл с расширением чье сотрудничество вы хотите изменить, а затем нажмите Открыть с.
- В Открыть с помощью диалоговое окно, выберите программу ти котором вы хотите, чтобы открыть файл, или нажмите Обзор, чтобы найти программу, которую вы хотите.
- Выберите Всегда использовать выбранную программу, чтобы открыть такой файл флажок.
Поддерживаемые операционные системы
Windows Server 2003/2008/2012/2016, Windows 7, Windows 8, Windows 10, Linux, FreeBSD, NetBSD, OpenBSD, Mac OS X, iOS, Android
Формат FASTQ представляет собой текстовый формат для хранения как биологической последовательности (обычно нуклеотидной последовательности ) и его соответствующие показатели качества. И буква последовательности и показатель качества, каждый кодируются с помощью одного ASCII символа для краткости.
содержание
Формат
Файл FASTQ обычно использует четыре линии на последовательность.
- Строка 1 начинается с символа «@» и сопровождается идентификатором последовательности и дополнительным описанием (как FASTA строке заголовка).
- Строка 2 сырые буквы последовательности.
- Строка 3 начинается с «+» характером и необязательно следует тем же идентификатор последовательности (и любое описание) еще раз.
- Строка 4 кодирует значение качества для последовательности в строке 2, и должна содержать одинаковое количество символов в виде букв в последовательности.
Файл FASTQ, содержащий одну последовательность может выглядеть следующим образом:
Байты , представляющего качество работает от 0x21 (самого низкого качества, в ASCII «!») Для 0x7E ( сам высокого качество; «
» в ASCII). Вот значение качества символов слева направо порядке возрастания качества ( ASCII ):
Оригинальные Sanger FASTQ файлы также позволили последовательности и качества строка должна быть обернута (раскол на несколько строк), но это, как правило, не рекомендуются, поскольку это может сделать разбор сложным из-за неудачный выбор «@» и «+» в качестве маркеров (эти символы могут также возникать в строке качества).
идентификаторы последовательности Illumina
Последовательности из Illumina программного обеспечения используют системный идентификатор:
С Casava 1.8 формат строки «@» изменилась:
Обратите внимание, что более поздние версии программного обеспечения Illumina вывода количество образца (как взяты из образца листа) вместо индекса последовательности. Например, следующий заголовок может появиться в первом образце партии:
NCBI Последовательность чтения Архив
FASTQ файлы из INSDC Последовательность чтения архива часто включают в себя описание, например ,
В этом примере есть NCBI назначенный идентификатор, и описание имеет оригинальный идентификатор из Solexa / Illumina (как описано выше) , плюс длина чтения. Секвенирование проводили в паре-концевой режиме (
500bp размера вставки), см SRR001666 . Формат вывода по умолчанию fastq-дамп производит целые пятна, содержащее любые технические операции чтения и , как правило , одиночные или парный-конец биологические читают. Его поведение было обусловлено требованиями нескольких проектов , действующих на время fastq-свалка была разработана, прежде всего 1000 геномов проекта .
Современное использование FASTQ почти всегда включает в себя расщепление места в его биологический читает, как описано в подателе предоставленных метаданных:
Когда присутствует в архиве, fastq-свалка может попытаться восстановить чтение имен исходного формата. Следует отметить, что NCBI не сохраняет исходные имена чтения по умолчанию:
В приведенном выше примере, были использованы оригинальные имена чтения, а не accessioned чтения имени. NCBI присоединений пробегов и читает они содержат. Оригинальные имена чтения, назначенные секвенсорами, способны функционировать как локально уникальные идентификаторы для чтения, и передать ровно столько информации, сколько серийный номер. Идентификаторы выше были алгоритмически назначены на основании выполнения информации и геометрических координат. Ранний SRA погрузчики разобраны эти идентификаторы и хранят их разлагают компоненты внутри. NCBI остановил запись чтения имен, потому что они часто редактировались исходным формат вендоров для того, чтобы связать некоторую дополнительную информацию, значимую для конкретного трубопровода обработки, и это вызвало нарушение в формате имя, которое привело к большому числу отвергнутых представлений. Без четкой схемы для имен чтения, их функция остается то, что из уникального идентификатора для чтения, передавая тот же объем информации, как прочитать серийный номер.
Также отметим , что fastq-дампа преобразует эти данные FASTQ из исходного Solexa / Illumina кодирования к стандарту Sanger (см кодировки ниже). Это происходит потому , что SRA служит хранилищем информации NGS, а не в формате. Различные * -dump инструменты способны производить данные в нескольких форматах из одного источника. Требования , предъявляемые к этом было продиктованы пользователями в течение нескольких лет, с большинством раннего спроса , поступающим из геномов проекта 1000 .
вариации
Качественный
Трубопровод Solexa (то есть, программное обеспечение поставляется с Illumina Genome Analyzer) ранее использовали различные отображения, кодирующие коэффициенты р / (1- р ) вместо вероятности р :
кодирование
Альтернативная интерпретация этого ASCII кодировки была предложена. Кроме того, в Illumina работает с помощью элементов управления PhiX, наблюдался символ «B», чтобы представить «неизвестный показатель качества». Частота ошибок «B» считывает был примерно 3 Phred баллов снизить средний балл наблюдали в данном цикле.
- Начиная с Illumina 1.8, оценки качества были в основном вернулись к использованию формата Sanger (Phred + 33).
Для получения сырья читает, диапазон баллов будет зависеть от технологии и базовой вызывающей используется, но как правило, будет до 41 для недавней химии Illumina. Поскольку максимальный балл наблюдаемого качества было ранее только 40, различные сценарии и инструменты ломаются, когда они сталкиваются с данными с качеством значения больше, чем 40. Для обрабатываются чтениями, оценки могут быть еще выше. Например, значения качества из 45 наблюдаются в чтение из Лонг Read секвенирования Illumina службы (в ранее Moleculo).
Цветовое пространство
Для SOLiD данных, последовательность в цветовом пространстве, за исключением первой позиции. Значения качества являются те формата Sanger. Выравнивание инструментов различаются по предпочитаемой версии значений качества: некоторые включают в себя оценку качества ( «!» Установлен в 0, то есть) для ведущего нуклеотида, а другие нет. Последовательность чтения архив включает в себя это показатель качества.
моделирование
FASTQ чтение моделирование было приближено несколько инструментов. Сравнение этих инструментов можно увидеть здесь.
компрессия
счета значения качества примерно половина требуемого дискового пространства в формате FASTQ (перед сжатием), и, следовательно, сжатие значений качества может значительно снизить требования к хранению и ускорить анализ и передачу данных секвенирования. И без потерь и сжатие с потерями в последнее время рассматриваются в литературе. Например, алгоритм QualComp выполняет сжатие с потерями со скоростью (число бит на значение качества), указанной пользователем. На основании результатов теории скорость-искажение, он выделяет количество битов таким образом, чтобы свести к минимуму MSE (среднеквадратичной ошибки) между оригиналом (несжатый) и восстановленные значения качества (после сжатия). Другие алгоритмы сжатия значений качества включают SCALCE и Fastqz. Оба алгоритм сжатия без потерь, которые обеспечивают дополнительный контролируемый подход с потерями преобразования. Например, SCALCE уменьшает размер алфавита, основанного на наблюдении, что значения качества «соседних» аналогичны в целом. Для ориентира, см.
По состоянию на HiSeq 2500 Illumina дает возможность для вывода свойств, которые были крупнозернистые в качественные бункера. В Binned оценки вычисляются непосредственно из эмпирического показатель качества таблицы, которая сама по себе связана с оборудованием, программным обеспечением и химии, которые были использованы в ходе эксперимента секвенирования.
Расширение файла
Там нет стандартного расширения файла для файла FASTQ, но .fq и .fastq, которые обычно используются.
преобразователи форматов
- Biopython версия 1,51 года (interconverts Sanger, Solexa и Illumina 1.3+)
- EMBOSS версия 6.1.0 патч 1 года (interconverts Sanger, Solexa и Illumina 1.3+)
- BioPerl версии 1.6.1 и выше (interconverts Sanger, Solexa и Illumina 1.3+)
- BioRuby версия 1.4.0 и выше (interconverts Sanger, Solexa и Illumina 1.3+)
- BioJava версия 1.7.1 и выше (interconverts Sanger, Solexa и Illumina 1.3+)
- MAQ может конвертировать из Solexa в Sanger (использовать этот патч для поддержки Illumina 1.3+ файлов).
- fastx_toolkit Прилагаемый fastq_quality_converter программа может конвертировать Illumina в Sanger
преобразования командной строки
FASTQ в формате FASTA:
Illumina FASTQ 1,8 до 1,3
Illumina FASTQ 1,3 до 1,8
Illumina FASTQ 1.8 сырья качества для Binned качества (HiSeq QTable 2.10.1, HiSeq 4000)
Illumina FASTQ 1.8 сырья качества для clinto формата (визуальный блок представление)
Программа FastQC позволяет проводить быстрый и несложный контроль качества "сырых" данных секвенирования. Она проводит набор стандартных статистических анализов, которые можно использовать для предварительной оценки релевантности данных и выявления возможных проблем в полученной последовательности.
Для проведения анализа качества чтений была иcпользована FastQC, установленная на Kodomo. Для удобства работы я также установила на свой компьютер версию с графическим интерфейсом.
Использованная команда: fastqc chr11.fastq
В результате был получен архив chr11_fastqc.zip, содержащий отчет о работе программы в виде html файла - chr11_fastqc.html.
В целом качество чтений можно назвать уловлетворительным, однако для повышения качества лучше провести чистку.
Очистка чтений
Очистка чтений была проведена с помощью программы Trimmomatic. Адаптеры в исследуемой последовательности были уже удалены. Необходимо было отсечь с конца чтения нуклеотиды с неудовлетворительным качеством ( 5%, выдается Warning, >20% - Failure. Причиной ошибок в этом модуле обычно служит общее низкое качество проведенного секвенирования.
В нашей последовательности как до, так и после чистки, неопределенных оснований не встречается, что, безусловно, очень хорошо.
Картирование чтений
Чтения были откартированы с помощью программы BWA - Burrows-Wheeler Alignment Tool. Использованные команды указаны в таблице.
| Команда | Назначение | Результат |
|---|---|---|
| bwa index chr11.fasta | Индексирование референсной последовательности | Индексированный файл chr11.fasta |
| bwa mem chr11.fasta chr11_trim.fastq > chr11.sam | Выравнивание очищенных чтений с референсной последовательностью | Файл chr11.sam, содержащий выравнивание в формате SAM (Sequence Alignment/Map) |
Анализ выравнивания
Для анализа полученного выравнивания применялась программа Samtools, предназначенная для обработки файлов в формате SAM. Использованные команды указаны в таблице.
| Команда | Назначение | Результат |
|---|---|---|
| samtools view chr11.sam -b -o chr11.bam | Перевод выравнивания в бинарный формат .bam (опция -b меняет формат выходного файла с установленного по умолчанию, -o обозначат имя выходного файла). | chr11.bam |
| samtools sort chr11.bam -T smth.txt -o chr11_sort.bam | Сортировка выравнивания чтений с референсом по координате в референсе начала чтения (опция -T позволяет записывать временные файлы в файл smth.txt, а не в stdout). | chr11_sort.bam |
| samtools index chr11_sort.bam | Индексирование отсортированного выравнивания | Индексированный chr11_sort.bam |
| samtools idxstats chr11_sort.bam > reads.out | Выяснение числа чтений, откартированных на геном | reads.out |
Из файла reads.out, я получила информацию, что на хромосому откартировалось 4062 чтения и 2 чтения откартированы не были.
Определение среднего покрытия экзона
Для начала было вычислено покрытие для каждого из нуклеотидов и был выбран один из нуклеотидов с наилучшим покрытием (185). Он имел координату 116628513. Данный нуклеотид был найден в Genome Browser. Выяснилось, что он принадлежит гену BUD13 c координатами complement(116618886..116643714) и длиной 24829. Экзон, содержащий данный нуклеотид, имеет границы 116628481-116628666.
 |
| Рис. 21. Ген BUD13 |
 |
| Рис. 22. Экзон, содержащий нуклеотид 116628513 |
Использованные команды:
| Команда | Назначение | Результат |
|---|---|---|
| samtools depth chr11_sort.bam > depth.out | Вычисление покрытия каждого нуклеотида | depth.out |
| samtools depth -r chr11:116628481-116628666 chr11_sort.bam > exon.out | Вычисление покрытия нуклеотидов в границах экзона | exon.out |
Исходя из данных файла exon.out с помощью Excel было вычислено среднее покрытие выбранного экзона. Оно составило 143.48, что является достаточно хорошим показателем.
Поиск SNP и инделей
| Команда | Назначение | Результат |
|---|---|---|
| samtools mpileup -uf chr11.fasta chr11_sort.bam -o snp.bcf | Создание файла с полиморфизмами в формате .bcf | snp.bcf |
| bcftools call -cv snp.bcf -o snp.vcf | Создание файла со списком отличий между референсом и чтениями в формате .vcf | snp.vcf |
Всего в файле snp.vcf указано 23 полиморфизма, из них 1 вставка, 1 делеция и 21 замена. Покрытие и качество полиморфизмов встречается как хорошее (например качество 188.009 и покрытие 92), так и не очень (например качество 4.76921 и покрытие 2). Среднее значение качества - 118,82, покрытия - 23,56. В целом показатели достаточно нормальные.
| Примеры полиморфизмов | |||||
|---|---|---|---|---|---|
| Координата | Тип | В референсе | В прочтении | Качество прочтения на участке | Глубина покрытия на участке |
| 116628401 | Замена | T | С | 188.009 | 92 |
| 116650454 | Делеция | ATCTCT | ATCT | 196.468 | 22 |
| 116657590 | Вставка | CAAA | CAAAA | 144.467 | 63 |
Аннотация SNP
Далее необходимо было аннотировать полученные SNP с помощью программы ANNOVAR. Для этого сначала нужно было подготовить входной файл, чтобы программа моглас ним работать. В первую очередь из файла snp.vcf были удалены все индели, полученный файл - snp_noindel.vcf. Затем был использован скрипт convert2annovar.pl.
| Команда | Назначение | Результат |
|---|---|---|
| perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 snp_noindel.vcf -outfile snp.avinput | Подготовка входного файла | snp.avinput |
Полученный файл использовался для аннотации SNP по базам данных refgene, dbsnp, 1000 genomes, GWAS и Clinvar c помощью скрипта annotate_variation.pl.
В Annovar существуют 3 типа аннотаций по базам данных, основанных на: генной разметке (gene-based annotation); разметке других регионов генома (region-based annotation); фильтрации (filter-based annotation). Тип использованной аннотации указан для каждой базы данных отдельно.
Refgene
Тип аннотации: gene-based annotation.
Использованная команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -out chr11.refgene -build hg19 snp.avinput /nfs/srv/databases/annovar/humandb/
-
- содержит описание всех полиморфизмов. - содержит описание полиморфизмов внутри экзонов.
- chr11.refgene.log - содержит отчет о работе команды.
- exonic - полиморфизм внутри экзона (частично или полностью)
- splicing - полиморфизм в пределах 2 bp от границы сплайсинга (число bp можно изменить)
- ncRNA - полиморфизм полностью или частично входит в транскрипт, не имеющий аннотации как кодирующий
- UTR5 - полиморфизм полностью или частично входит в 5′-нетранслируемую область
- UTR3 - полиморфизм полностью или частично входит в 3′-нетранслируемую область
- intronic - полиморфизм полностью или частично внутри интрона
- downstream - полиморфизм в пределах 1-kb downstream от сайта окончания транскрипции (параметр может быть изменен)
- upstream - полиморфизм в пределах 1-kb upstream от сайта начала транскрипции (параметр может быть изменен)
- intergenic - полиморфизм на пересечении генов
Если полиморфизм попадает в категории exonic/intronic/ncRNA, то во второй колонке того же файла указывается имя соответсвующего гена (если это сразу несколько генов, то они разделяются запятой). Если полиморфизм попадат в какую-либо другую категорию, то во второй колонке будут указаны два соседних гена и расстояние между ними.
В моем случае в категории, для которых указывается ген, попали 17 полиморфизмов. Все они расположены на одном из трех генов - KCNJ11, BUD13 или ZPR1.
Ген KCNJ11 кодирует белок потенциалозависимого калиевого ионного канала. В нем оказалось 2 полиморфизма категории exonic.
Ген BUD13 является белок кодирующим, однако функция его в NCBI не прописана. В нем оказалось 7 полиморфизмов категории intronic и 2 - exonic.
Ген ZPR1 кодирует белок, который взаимодействует с белком выживания двигательных нейронов (survival motor neuron protein - SMN1), усиливая сплайсинг пре-мРНК и активируя нейронную дифференцировку и рост аксонов. В нем оказалось 5 полиморфизмов категории intronic и 1 полиморфизм категории exonic.
Большинство аннотированных полиморфизмов (57%) принадлежат категории intronic, и лишь 5 (23%) - категории exonic. Это можно считать достаточно логичным, так как замены в некодирующих последовательностях не подвергаются такому тщательному отбору, как замены в последовательностях, кодирующих белки. Полиморфизмы категории exonic могут приводить к аминокислотным заменам и, в случае несинонимичных замен, к нарушению функии белка.
В таблице более подробно рассмотрены полиморфизмы, попавшие внутрь экзонов, с указанием нуклеотидных и аминокислотных замен:
| № | Координаты (*) | Ген | Н. замена | Тип замены | Синонимичность | Качество чтений | Глубина покрытия | А.к. замена |
|---|---|---|---|---|---|---|---|---|
| 1 | 17408630 | KCNJ11 | C > T | гомозиготная | нет | 221,999 | 35 | V250I, V337I |
| 2 | 17409572 | KCNJ11 | T > C | гомозиготная | нет | 138,133 | 8 | K23E |
| 3 | 116633825 | BUD13 | C > T | гетерозиготная | да | 194,009 | 33 | P160P |
| 4 | 116633862 | BUD13 | G > A | гетерозиготная | нет | 224,009 | 40 | P148L |
| 5 | 116655600 | ZPR1 | G > A | гетерозиготная | нет | 225,009 | 37 | A264V |
- Полиморфизм 1: общий вид, увеличение
- Полиморфизм 2: общий вид, увеличение
- Полиморфизм 3: общий вид, увеличение
- Полиморфизм 4: общий вид, увеличение
- Полиморфизм 5: общий вид, увеличение
Dbsnp
Тип аннотации: filter-based annotation.
Использованная команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr11.dbsnp -build hg19 -dbtype snp138 snp.avinput /nfs/srv/databases/annovar/humandb/
-
- содержит полиморфизмы, имеющие rs в dbsnp, то есть аннотированные в этой базе данных. - содержит отфильтрованные полиморфизмы (не имеющие rs в dbsnp).
- chr11.dbsnp.log - содержит отчет о работе команды.
1000 genomes
Тип аннотации: filter-based annotation.
Использованная команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr11.1000 -buildver hg19 -dbtype 1000g2014oct_all snp.avinput /nfs/srv/databases/annovar/humandb/
-
- содержит полиморфизмы, имеющие rs в 1000 genomes, и их частоты. - содержит полиморфизмы, не имеющие rs в 1000 genomes.
- chr11.1000.log - содержит отчет о работе команды.
| № | Координата SNP | Ген | Частота |
|---|---|---|---|
| 1 | 17408630 | KCNJ11 | 0,730631 |
| 2 | 17409572 | KCNJ11 | 0,737021 |
| 3 | 116633825 | BUD13 | 0,545128 |
| 4 | 116633862 | BUD13 | 0,088858 |
| 5 | 116655600 | ZPR1 | 0,046725 |
Видно, что найденные нами экзонные полиморфизмы встречаются достаточно часто, для 3 из 5 snp вероятность превышает 50%.
Тип аннотации: region-based annotation.
Использованная команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -out chr11.gwas -build hg19 -dbtype gwasCatalog snp.avinput /nfs/srv/databases/annovar/humandb/
-
- содержит snp, имеющие известное клиническое значение.
- chr11.gwas.log - содержит отчет о работе команды.
| № | Координата | Ген | Клиническое значение |
|---|---|---|---|
| 1 | 17408630 | KCNJ11 | Диабет 2-го типа |
| 2 | 17409572 | KCNJ11 | Диабет 2-го типа |
| 4 | 116633862 | BUD13 | Метаболический синдром |
Clinvar
Тип аннотации: filter-based annotation.
Использованная команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr11.clincar -buildver hg19 -dbtype clinvar_20150629 snp.avinput /nfs/srv/databases/annovar/humandb/
-
- содержит snp, аннотированные в Clinvar. - содержит snp, не аннотированные в Clinvar.
- chr11.clincar.log - содержит отчет о работе команды.
Сводная таблица SNP
Сводную таблицу Excel можно скачать по ссылке.
Использовались только файлы с анноированными snp (dropped).
К написанию данной статьи меня побудил вопрос на Тостере, связанный с WSL. Я, после нескольких лет использования систем на ядре Linux, около полугода назад перешел к использованию Windows 10 на домашнем ПК. Зависимость от терминала и Linux окружения в моей работе практически сразу привели меня к вопросу: или ставить виртуалку или попробовать WSL. Я выбрал второе, и остался вполне доволен.
Под катом я расскажу как установить и настроить WSL, на какие я наткнулся проблемы и ограничения, как запускать Linux приложения из Windows и наоборот, а так же как интегрировать элементы окружения Xfce в окружение рабочего стола Windows.

Никогда не думал, что однажды вернусь на Windows, но повод попробовать мне дали стечения обстоятельств: жена, далекая от IT, дергала почти каждый раз, когда у нее возникала необходимость воспользоваться компом; проснулась ностальгия по одной игре, но она никак не хотела адекватно работать под wine; а тут еще мне подарили коробочную Windows 10 Pro. WSL я поставил чуть ли не сразу после установки системы, поигрался несколько вечеров, понял, что продукт для моих задач годный, но хочется более привычный терминал и вообще некоторых удобств.
Установка WSL и дистрибутива
Сразу оговорюсь, в интернете можно найти описание установки с помощью выполнения команды lxrun /install в командной строке или консоли PowerShell. Данный способ больше не работает (после выхода WSL в стабильный релиз). Насколько мне известно, сейчас WSL можно установить только из Microsoft Store вместе с предпочитаемым дистрибутивом.
Так же отмечу, что когда установку производил я, на выбор были доступны дистрибутивы OpenSUSE, SUSE Linux Enterprise и Ubuntu 16.04 — последний я и установил. Сейчас также доступны Ubuntu 18.04, Debian 9 и Kali Linux, возможно появятся и другие дистрибутивы. Действия по установке могут отличаться. Так же, часть проблем описанных в статье может быть уже исправлена.
Находим в магазине желаемый дистрибутив и устанавливаем. Установка пройдет быстро, так как скачает только эмулятор ядра Linux и утилиту для запуска подсистемы, которая окажется в системной папке в трех экземплярах: wsl.exe, bash.exe и ubuntu.exe (вместо ubuntu будет имя Вашего дистрибутива). Все они равнозначны и делают одно и то же — запускают собственный эмулятор терминала, в нем linux'овый bash работающий под эмулятором ядра. При первом же запуске нас попросят придумать логин и пароль для пользователя по умолчанию, а после произойдет непосредственно установка дистрибутива. В качестве пользователя по умолчанию указываем root без пароля — это потребуется для дальнейших шагов. Безопасность не пострадает, кроме того при подготовке материалов к статье, в англоязычном туториале, я наткнулся на информацию, что новые версии WSL теперь делают пользователем по умолчанию root без пароля без лишних вопросов.
Дожидаемся установки. Далее первым делом стоит обновить зеркала apt на ближайшие. Для этого понадобится CLI текстовый редактор. В комплекте только vi, я же больше предпочитаю nano, поэтому ставлю его:
sudo вводить не требуется, так как мы уже под root'ом. Отредактируем файл /etc/apt/sources.list:
У меня лучше всего работают зеркала Яндекса, поэтому мой файл выглядит так:
Нажимаем Ctrl+O для сохранения и Ctrl+X для выхода. Теперь можно обновить систему до актуального состояния:
После обновления можно создать нашего основного пользователя. В данной статье я назову его user1, Вы же можете задать привычное имя:
Далее переходим в папку юзера, зайдем под ним, установим пароль и отредактируем файл
Все, подсистема готова к использованию… почти.
Установка X-сервера, Xfce и прочих GUI'шных приложений
Первая же проблема, на которую я натолкнулся — bash-completion в предлагаемом эмуляторе терминала работал, мягко говоря, некорректно. Кроме того, данный эмулятор не умеет вкладки, а каждый его экземпляр запускает все в новом пространстве процессов, с отдельным init'ом (который кстати не заменить). Мне захотелось нормальный эмулятор терминала, некоторых других GUI приложений, а так же панельку, чтоб это все быстро запускать.
Когда я гуглил этот вопрос, я наткнулся на множество проблем, вроде необходимости перевода dbus на tcp протокол. На данный момент всех этих проблем нет. В подсистеме нормально работают unix-domain-socket'ы и все спокойно общается через них.
Первым делом нам понадобится X-сервер, притом установленный в основную систему (в Windows). Лично я использую для этих целей VcXsrv — порт X11 на Windows. Официальный сайт указанный в about самой утилиты его сейчас не предоставляет, поэтому гуглим установщик и устанавливаем все по умолчанию.
Пока идет установка возвращаемся в терминал WSL, командой exit выходим обратно в root'а. Первым делом настроим русские локали:
Далее установим некоторые компоненты Xfce. Можно конечно установить его целиком из мета-пакета, но большинство компонентов нам не понадобится, а модульная архитектура Xfce позволяет нам поставить только необходимое:
Запускать каждый раз окружение руками не очень удобно, поэтому я автоматизировал данный процесс. Для этого в основной системе создадим в удобном для нас месте папку, а в ней 3 файла для запуска:
-
config.xlaunch — файл настроек для VcXsrv
x-run.vbs — WSL всегда запускается со своим эмулятором терминала, если его закрыть — завершатся все его дочерние процессы. Чтоб данное окно не мозолило глаза, неплохо его запускать скрытым. К счастью в Windows встроен интерпретатор VBScript, который позволяет это сделать в одну строчку:
Поясню, что здесь происходит. Мы говорим VBscript выполнить приложение wsl с параметром cd /home/user1; DISPLAY=:0 LANG=ru_RU.UTF-8 su user1 -c xfce4-session , папка запуска нам не важна, поэтому пустая строка, действие open — запуск, 0 — скрытый режим. Самому wsl мы отдаем команду на выполнение: переход в папку пользователя, затем с установкой переменных окружения DISPLAY (дисплей X-сервера) и LANG (используемая локаль) мы запускаем xfce4-session от имени нашего пользователя user1 (благодаря команде su)
Далее можем запустить наш start.bat и настроить панель Xfce под себя. Замечу, что здесь я наткнулся на еще одну проблему — панель прекрасно отображается поверх всех окон, но вот выделить себе место, как панель на рабочем столе Windows она не может. Если кто знает решение данной проблемы, поделитесь в комментариях.
Ну и под конец данной части, скриншот моего рабочего стола:

Взаимодействие окружения Windows и окружения подсистемы Linux
Запускать Linux приложения напрямую из Windows можно через те же 3 команды — bash, wsl или ubuntu. Не забываем, что по умолчанию запуск идет от root, поэтому стоит понижать привилегии через su , так же нужно не забывать передавать переменную окружения DISPLAY=:0 если приложению требуется X-сервер. Так же нужно менять папку, из которой должно работать приложение, через cd внутри WSL. Пример, посчитаем md5 для file.txt на диске D средствами Linux'овой md5sum:
Доступ к файловой системе Linux так же имеется, лежит она в %localappdata%\Packages\CanonicalGroupLimited.UbuntuonWindows_79rhkp1fndgsc\LocalState\rootfs . Читать таким образом файлы можно, а вот писать — не желательно, можно поломать файловую систему. Думаю проблема в том, что Windows не умеет работать с правами и владельцами файловой системы Linux.
Из Linux так же можно запускать Windows приложения. Просто запускаем exe-шник и он выполнится в основной системе.
Диски Windows монтируются в /mnt в соответствии со своими буквами в нижнем регистре. Например диск D будет смонтирован в /mnt/d . Из Linux можно свободно читать и писать файлы Windows. Можно делать на них симлинки. Права у таких файлов всегда будут 0777, а владельцем будет root.
Сетевой стек у подсистемы общий с Windows. Сервер поднятый в Linux будет доступен на localhost в Windows и наоборот. Однако unix-domain-socket для Windows будет просто пустым файлом, работать с этим можно только внутри Linux. Выход во внешнюю сеть у Linux так же есть, в том числе можно слушать порты, если этого не запрещает фаервол.
ifconfig в Linux и ipconfig в Windows выдают одинаковую информацию о сетевых интерфейсах.
Из диспетчера задач Windows можно спокойно прибить процесс внутри подсистемы Linux. Однако Linux увидит только свои процессы.
Особенности, ограничения и подводные камни
Ядро Linux в WSL не настоящее. Это всего лишь прослойка-эмулятор, которая часть Linux-специфичных задач выполняет сама, а часть проксирует напрямую в ядро winNT. Большая часть api в нем реализована, но не все. Свое ядро собрать не получится, как и не получится подключить модули ядра (.ko, Kernel Object).
Init процесс у WSL тоже свой и заменить его, например, на system.d не выйдет. У меня давно есть желание написать менеджер демонов на go, который бы работал с файлами юнитов system.d и предоставлял бы схожий интерфейс, да все руки не доходят.
Нет поддержки openFUSE, соответственно примонтировать виртуальную или удаленную файловую систему не получится. Так же нельзя сделать mount из файла, mount вообще ничего кроме bind здесь, похоже, не умеет.
Так же нет никакой возможности разбить файловую систему Linux на несколько разделов/дисков.
Прямой доступ к железу практически отсутствует. Все таки мы находимся в песочнице Windows, а не в полноценном Linux. /dev и /sys заметно пустуют, в них лишь проц да виртуальные устройства. Доступ к GPU — только через X-сервер, напрямую — никак, так что нейросети обучать придется в Windows.
В JS разработке столкнулся с тем, что electron.js отказался запускаться в WSL, пришлось дублировать окружение node.js в Windows.
Итоги
Статья получилась довольно длинной, надеюсь, что она окажется еще и полезной.
WSL для меня лично оказался инструментом вполне юзабельным, решающим мои задачи fullstack backend разработчика. Виртуалка с Linux за полгода так и не понадобилась. По общим ощущениям Windows+WSL намного функциональнее, чем Linux+Wine.
Пока писал статью, обнаружил, что в Microsoft Store появилась сборка WSL с Debian 9.3, данный дистрибутив мне более симпатичен, чем Ubuntu, поэтому буду пробовать ставить.
Программы, которые поддерживают FASTQ расширение файла
Следующий список содержит программы, сгруппированные по 3 операционным системам, которые поддерживают FASTQ файлы. Файлы с суффиксом FASTQ могут быть скопированы на любое мобильное устройство или системную платформу, но может быть невозможно открыть их должным образом в целевой системе.
Программы, обслуживающие файл FASTQ
Updated: 02/05/2020
Как открыть файл FASTQ?
Причин, по которым у вас возникают проблемы с открытием файлов FASTQ в данной системе, может быть несколько. Что важно, все распространенные проблемы, связанные с файлами с расширением FASTQ, могут решать сами пользователи. Процесс быстрый и не требует участия ИТ-специалиста. Приведенный ниже список проведет вас через процесс решения возникшей проблемы.
Шаг 1. Установите Sublime Text программное обеспечение

Основная и наиболее частая причина, препятствующая открытию пользователями файлов FASTQ, заключается в том, что в системе пользователя не установлена программа, которая может обрабатывать файлы FASTQ. Наиболее очевидным решением является загрузка и установка Sublime Text или одной из перечисленных программ: PSPad, NotePad++ text editor, Atom. Полный список программ, сгруппированных по операционным системам, можно найти выше. Если вы хотите загрузить установщик Sublime Text наиболее безопасным способом, мы рекомендуем вам посетить сайт Sublime HQ Pty Ltd и загрузить его из официальных репозиториев.
Шаг 2. Проверьте версию Sublime Text и обновите при необходимости

Вы по-прежнему не можете получить доступ к файлам FASTQ, хотя Sublime Text установлен в вашей системе? Убедитесь, что программное обеспечение обновлено. Может также случиться, что создатели программного обеспечения, обновляя свои приложения, добавляют совместимость с другими, более новыми форматами файлов. Это может быть одной из причин, по которой FASTQ файлы не совместимы с Sublime Text. Самая последняя версия Sublime Text обратно совместима и может работать с форматами файлов, поддерживаемыми более старыми версиями программного обеспечения.
Шаг 3. Свяжите файлы FASTQ Format с Sublime Text
После установки Sublime Text (самой последней версии) убедитесь, что он установлен в качестве приложения по умолчанию для открытия FASTQ файлов. Метод довольно прост и мало меняется в разных операционных системах.
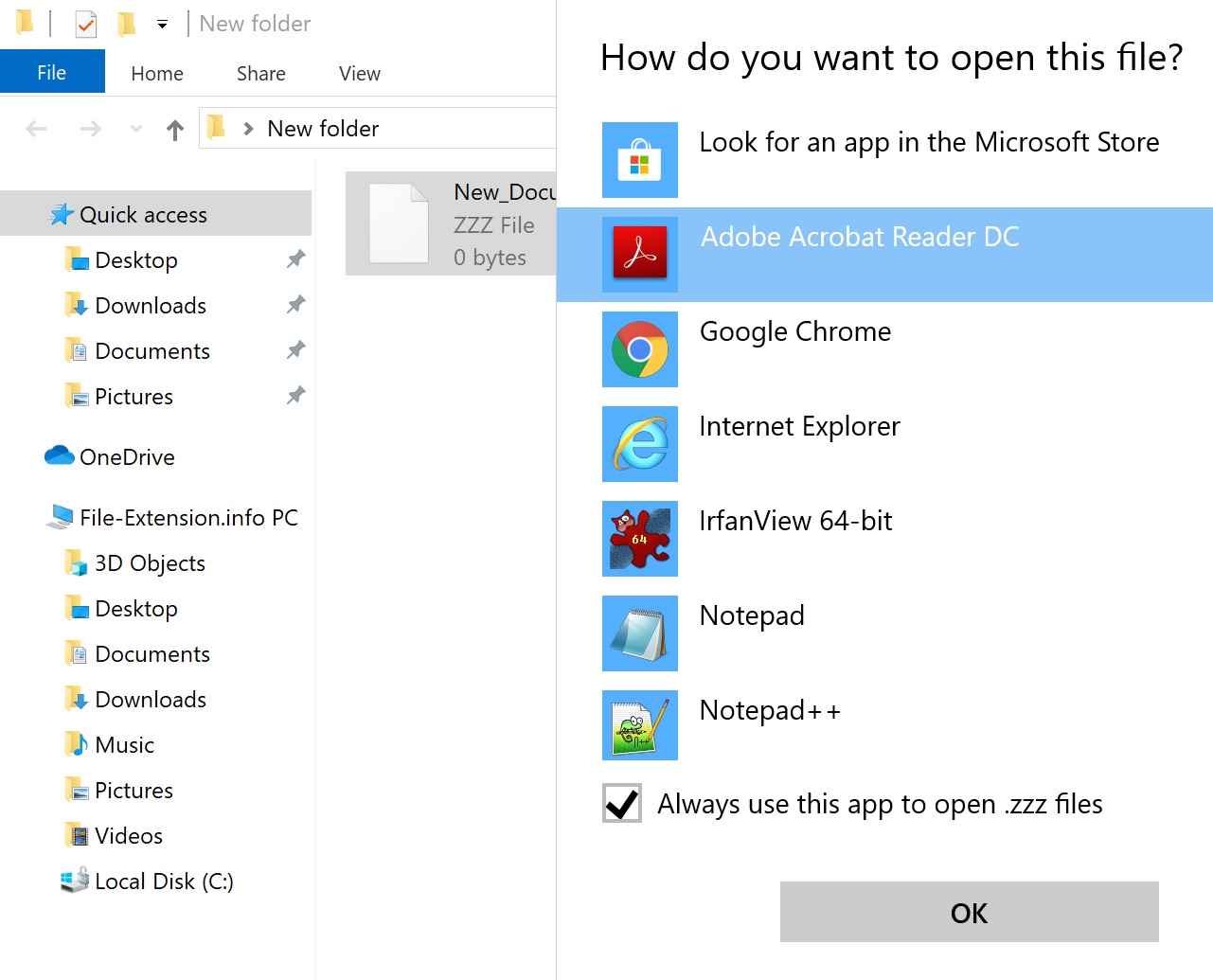
Изменить приложение по умолчанию в Windows
- Нажатие правой кнопки мыши на FASTQ откроет меню, из которого вы должны выбрать опцию Открыть с помощью
- Выберите Выбрать другое приложение → Еще приложения
- Чтобы завершить процесс, выберите Найти другое приложение на этом. и с помощью проводника выберите папку Sublime Text. Подтвердите, Всегда использовать это приложение для открытия FASTQ файлы и нажав кнопку OK .
Изменить приложение по умолчанию в Mac OS
Шаг 4. Убедитесь, что файл FASTQ заполнен и не содержит ошибок
Если проблема по-прежнему возникает после выполнения шагов 1-3, проверьте, является ли файл FASTQ действительным. Отсутствие доступа к файлу может быть связано с различными проблемами.

1. Убедитесь, что FASTQ не заражен компьютерным вирусом
Если случится так, что FASTQ инфицирован вирусом, это может быть причиной, которая мешает вам получить к нему доступ. Немедленно просканируйте файл с помощью антивирусного инструмента или просмотрите всю систему, чтобы убедиться, что вся система безопасна. Если сканер обнаружил, что файл FASTQ небезопасен, действуйте в соответствии с инструкциями антивирусной программы для нейтрализации угрозы.
2. Убедитесь, что файл с расширением FASTQ завершен и не содержит ошибок
Если вы получили проблемный файл FASTQ от третьего лица, попросите его предоставить вам еще одну копию. Возможно, что файл не был должным образом скопирован в хранилище данных и является неполным и поэтому не может быть открыт. При загрузке файла с расширением FASTQ из Интернета может произойти ошибка, приводящая к неполному файлу. Попробуйте загрузить файл еще раз.
3. Проверьте, есть ли у пользователя, вошедшего в систему, права администратора.
Существует вероятность того, что данный файл может быть доступен только пользователям с достаточными системными привилегиями. Войдите в систему, используя учетную запись администратора, и посмотрите, решит ли это проблему.
4. Убедитесь, что в системе достаточно ресурсов для запуска Sublime Text
Если в системе недостаточно ресурсов для открытия файлов FASTQ, попробуйте закрыть все запущенные в данный момент приложения и повторите попытку.
5. Убедитесь, что ваша операционная система и драйверы обновлены
Современная система и драйверы не только делают ваш компьютер более безопасным, но также могут решить проблемы с файлом FASTQ Format. Возможно, что одно из доступных обновлений системы или драйверов может решить проблемы с файлами FASTQ, влияющими на более старые версии данного программного обеспечения.
Вы хотите помочь?
Если у Вас есть дополнительная информация о расширение файла FASTQ мы будем признательны, если Вы поделитесь ею с пользователями нашего сайта. Воспользуйтесь формуляром, находящимся здесь и отправьте нам свою информацию о файле FASTQ.
Читайте также:
- Центр действий windows 10 как открыть
- Как загрузить последнюю удачную конфигурацию windows xp
- Linux команда find exec
- Dev или beta windows 11 что лучше
- Linux обход блокировки торрентов